An Efficient Synthesis of Enantiomerically Pure γ-Aminobutyric Acid (GABA) Derivatives ()

1. Introduction
GABA is the most common inhibitory neurotransmitters in the mammalian central nervous system (CNS) where it exerts its effects through inhibiting GABA receptors channels. And decreased GABA biological activity results in the excessive excitement of CNS (namely Neuronal Firing). The dysfunction of the central GABA system is responsible for the development and outbreak of epilepsy [1], Huntington’s, Parkinson’s diseases [2] and other psychiatric disorders, such as anxiety and pain [3]. Various GABA derivatives and analogs are widely recognized as CNS depressants, for example: Phenibut, Baclofen (Lioresal and Baclon®), Vigabatrin, Gabapentin (GBP, Neurontin), Pregabalin (Lyrica, Pfizer’s blockbuster drug, β-i-Bu-GABA) (Figure 1).
Enantiomers of a drug are often known to play an important role in pharmacological actions, pharmacokinetics, toxicities and metabolism. The biological activity of some drugs depends on their absolute configuration. Therefore, the use of a single isomer of a given drug is usually recommended for clinical use [4]. It has reported that (R)-enantiomer of GABA derivatives is much more active than (S)-enantiomer [5] [6] [7]. For example, in pharmacological tests of locomotor activity, antidepressant and pain effects, (S)-phenibut was inactive in doses up to 500 mg/kg, but in contrast, (R)-phenibut turned out to be two times more potent than racemic phenibut in most of the tests. In the forced swimming test, at a dose of 100 mg/kg only (R)-phenibut significantly decreased immobility time. Consequently, obtaining enantiomerically pure GABA derivatives as a new modulators has become one of the most effective approaches for the development of extracellular GABA homeostasis in the CNS.
In this paper, we report the asymmetric Michael addition of S, S’-diphenyldi- thiomalonate 2 to trans-β-nitroolefins 1, catalyzed by chiral cinchona alkaloid-derived thioureas organocatalysts 3, to give a series of corresponding chiral β-aryl-γ-nitroalkanes 4 in moderate to good yields, along with excellent stereoselectivities. And their transformations into biologically attractive chiral GABA derivatives 6 via reduction/cyclization/hydrolysis cascade reactions were reported [8].
2. Experimental
2.1. Materials
Trans-β-nitroolefins, benzotrifluoride, TiCl3 were purchased from commercial
![]()
Figure 1. GABA and its analogs used as CNS depressants.
suppliers and were used without further purification. S, S’-diphenyldithiomalo- nate 2 and cinchona alkaloid-derived thioureas 3 were previously synthesized in our laboratory. Zinc powder was freshly activated before usage. DCM and toluene were freshly distilled before usage. Analytical thin layer chromatography (TLC) was performed on Kiselgel 60 F254 plates. 1H NMR or 13C NMR spectra were recorded in CDCl3 and CD3OD, and the solvent signals (7.26/77.0 and 3.31/ 49.0 ppm) were used as reference. The yields are isolated by column chromatographygel plates. Percent enantiomeric excess (ee %) was determined by Agilent 1260 interfaced to a HP 71 series computer workstation with Chiralpak OD-H column. Optical rotations were determined on a polarimeter at 589 nm. Melting points were determined by using a XRC-1 microferrometer and are uncorrected. Specific rotations were measured in a Perkin-Elmer 343 polarimeter at room temperature and λ = 589 nm. The purification of all compounds was carried out by column chromatography using (silica gel 200 - 300).
2.2. Synthesis of β-Aryl-γ-Nitroalkanes 4 Vis Asymmetric Michael Addition
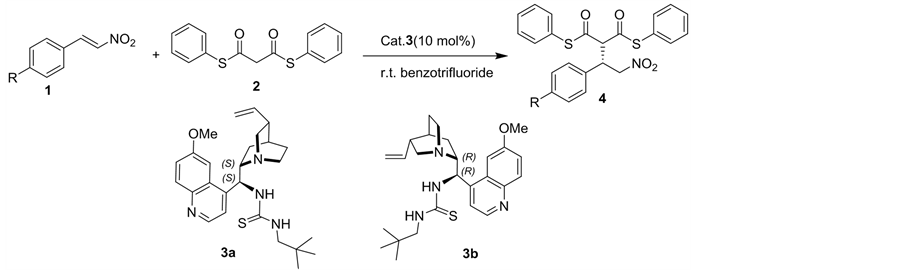
For the synthesis of the important intermediates β-aryl-γ-nitroalkanes 4, asymmetric Michael addition was studied [9]. We initially examined two kinds of cinchona alkaloid-derived bifunctional thioureas 3a-b as potential organocatalysts for the asymmetric Michael addition of S, S’-diphenyldithiomalonate 2 to trans-β-nitroolefins 1 in benzotrifuoride at room temperature, leading to the expected γ-nitroalkanes derivative 4 in good yield and excellent stereoselectivity (Table 1). Quinidine-derived thiourea 3b, pseudoenantiomer of 3a, effectively catalyzed the reaction and gave 4a, 4b and 4c with opposite absolute configuration in similar enantioselectivities (Table 1, entries 2, 4, 6 vs 1, 3, 5). Upon screening the various solvents, we found that benzotrifluoride produced better results than any other polar or less polar solvents, such as DCM, diethyl ether, touene and methanol.
Thus, the best reaction conditions was obtained when carrying out the reaction with 1.0 equiv of trans-β-nitroolefins 1 and 1.0 equiv of S, S’-diphenyldi- thiomalonate 2 in benzotrifluoride (1mL) at room tempreature in the presence of 10% mol of 3a/b as catalysts for 12 h (Table 1).
General experiment procedure: To the solution of trans-β-nitroolefins 1 (0.1 mmol) and S, S’-diphenyldithiomalonate 2 (0.1 mmol) in benzotrifluoride (1.0 m L) was added 3 (4.52 mg, 0.01 mmol). The resulting mixture was stirred at room temperature for 12 h until the reaction completed (monitored by TLC). After filtration, the products were furnished.
(S)-2-(2-Nitro-1-phenylethyl)-malonic acid diphenyl dithioester (4a). White solid; yield 92%; 96% ee; 1H NMR (400 MHz, CDCl3): d 7.47 - 7.41 (m, 5H), 7.39 - 7.33 (m, 6H), 7.28 - 7.26 (m, 2H), 7.14(dd, J = 7.60 Hz, J = 1.60 Hz, 2H), 4.88 - 4.82 (m, 2H), 4.48 (d, J = 8.80 Hz, 1H), 4.42-4.36 (m,1H) ppm; IR(KBr):ν 1697, 1670, 1558, 1473, 1438, 1377, 1261, 1022, 964, 798, 748, 686 cm−1; HPLC:
![]()
Table 1. The Asymmetric Michael Addition Reaction of S, S’-diphenyldithiomalonate 2 to trans-β-nitroolefins 1.
Chiralcel OD-H column, Hex/i-PrOH 70:30, 1.0 mL/min, t: major 15.6 min; t minor: 19.7 min.
(R)-2-(2-Nitro-1-phenylethyl)-malonic acid diphenyl dithioester (4a).White solid; yield 95%; 98% ee; 1H NMR and IR date are identical to (S)-4a; HPLC: Chiralcel OD-H column, Hex/i-PrOH 70:30, 1.0 mL/min, t major: 21.9 min; t: minor 19.0 min.
(S)-2-(1-(4-Methoxy-phenyl)-2-nitro-ethyl)-malonic acid diphenyl dithioester (4b). White solid; yield 90%; 89% ee; 1H NMR (CDCl3, 400 MHz): δ 7.43 - 7.35(m, 9H), 7.19-7.16(m, 4H), 6.87(d, J = 8.8 Hz, 2H), 4.81 - 4.79(m, 2H), 4.45(d, J = 9.6 Hz, 1H), 4.38 - 4.32(m, 1H), 3.79(s, 3H) ppm; IR(KBr): ν 1701, 1558, 1550, 1515, 1477, 1438, 1377, 1253, 1180, 1026, 964, 833, 748, 686 cm−1; HPLC: Chiralcel OD-H column, Hex/i-PrOH 70:30, 1.0 mL/min, t major: 18.2 min; t minor: 26.1 min.
(R)-2-(1-(4-Methoxy-phenyl)-2-nitro-ethyl)-malonic acid diphenyl dithioester (4b). White solid; yield 89%; 85% ee; 1H NMR and IR date are identical to (S)-5a; HPLC: Chiralcel OD-H column, Hex/i-PrOH 70:30, 1.0 mL/min, t major: 26.7 min; t minor: 18.4 min.
(S)-2-(1-(4- Chloro-phenyl)-2-nitro-ethyl)-malonic acid diphenyl dithioester (4c). White solid; yield: 91%; 75% ee; mp: 122˚C - 123˚C, [a]D25 = +77.6 (c = 0.25, CHCl3); 1H NMR (400 MHz, CDCl3): d 7.48 - 7.45 (m, 3H), 7.44 - 7.41 (m, 3H), 7.40 - 7.38 (m, 2H), 7.35 (d, J = 8.40 Hz, 2H), 7.22 (d, J = 8.40 Hz, 2H), 7.18 (dd, J = 7.60 Hz, J = 1.20 Hz, 2H), 4.81 (d, J = 5.60 Hz, 2H0, 4.44 (d, J = 9.60 Hz, 1H), 4.40 - 4 .35 (m, 1H) ppm; 13C NMR (100 MHz, CDCl3): δ 189.97, 189.15, 134.36, 133.97, 133.90, 133.47, 130.06, 129.92, 129.44, 129.32, 129.20, 129.06, 125.60, 125.54, 68.76, 43.36 ppm; IR(KBr):ν 1701, 1558, 1550, 1477, 1438, 1388, 1261, 1095, 1014, 960, 910, 829, 748, 686, 543 cm−1; HRMS-ESI (m/z): [M+Na]+ calcd for C24H18ClNNaO4S2 494.0258, found 494.0257; HPLC: Chiralcel OD-H column, Hex/i-PrOH 70:30, 1.0 mL/min, t major: 19.3 min; t minor: 30.1 min.
(R)-2-(1-(4-Chloro-phenyl)-2-nitro-ethyl)-malonic acid diphenyl dithioester (4c). Dark green solid; yield: 91%; 75% ee; mp: 122˚C - 123˚C, [a]D25 = +77.6 (c = 0.25, CHCl3); 1H NMR, 13C NMR, IR and HRMS-ESI date are identical to (S)-4c; HPLC: Chiralcel OD-H column, Hex/i-PrOH 70:30, 1.0 mL/min, t major: 24.1 min; t minor: 15.9 min.
2.3. Synthesis of (S)/(R)-β-Phenyl-γ-Lactam 5a
Considering that β-aryl-γ-nitroalkanes derivatives 4 are potential drug candidates, the transformation of adduct 4a to 5a was investigated as illustrated in Scheme 1. When adduct (S)-4a (96% ee) was treated with zinc/acetic acid and TiCl3, nitro group transformed into the amine and followed by intramolecular cyclization to produce the (S)-β-phenyl-γ-lactam 5a in good yield (90%) and only a few influence of enantioselectivity (91% ee) via subsequent reduction/ cyclization cascade reactions in a one-pot sequential protocol [10]. Under identical conditions, the adduct (R)-4a (98% ee) was transformed into (R)-β-phenyl- γ-lactam 5a in 90% yield and 89% ee (Scheme 1).
General procedure: To the solution of adduct 4a (218 mg, 0.5 mmol, 1.0 equiv) in AcOH (5.0 mL) was added freshly activated zinc powder (330 mg, 5 mmol, 10 equiv) and the resulting solution was stirred at 25˚C under a nitrogen atmosphere for 1h. After this period, TiCl3 (6.5 μL, 0.05 mmol, 0.1 equiv) was added and stirred for additional 1h until the reaction completed (monitored by TLC). The crude material was filtered, concentrated and purified by silicagel chromatography (petroleum ether/ethyl acetate = 10:1, v/v) to give the 5a as a white solid.
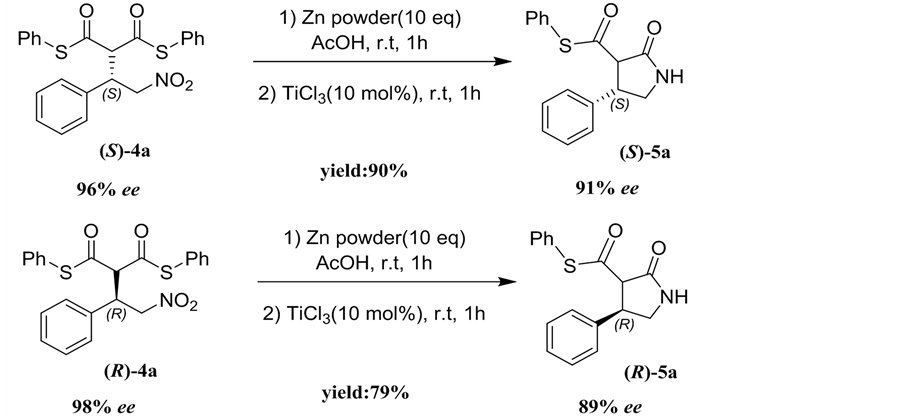
Scheme 1. Preparation of β-phenyl-γ-lactam 5a.
(S)-phenyl (4S)-2-oxo-4-phenylpyrrolidine-3-carbothioate (5a). White solid; mp: 160˚C - 162˚C; yield 90%; 91% ee; [a]D25 = +180 (c = 0.25, CHCl3); 1H NMR (400 MHz, CDCl3): δ 8.30 - 8.20 (s, 1H), 7.55-7.25 (m, 10H), 4.88 - 4.82 (m, 2H), 4.48 (d, J = 8.80 Hz, 1 H), 4.42 - 4.36 (m, 1H) ppm; IR (KBr): ν 3217, 3062, 2924, 2854, 1716, 1693, 1496, 1477, 1454, 1438, 1261, 1053, 1026, 1007, 821, 752 cm−1; HPLC: Chiralcel OD-H column, Hex/i-PrOH 70:30, 1.0 mL/min, t major: 23.0 min; t minor: 19.4 min.
(S)-phenyl (4R)-2-oxo-4-phenylpyrrolidine-3-carbothioate (5a). White solid; mp: 160˚C - 162˚C; yield 79%; 89% ee; [a]D25 = +210 (c = 0.25, CHCl3); 1H NMR and IR date are identical to (S)-5a; HPLC: Chiralcel OD-H column, Hex/i-PrOH 70:30, 1.0 mL/min, t major: 14.6 min; t minor: 18.8 min.
2.4. Synthesis of (S)/(R)-Phenibut
Phenibut is a therapeutically useful agonist of GABA type-B receptors and is used as a neuropsychotropic drug [11] [12]. To get the enantiomerically pure Phenibut, a tandem hydrolysis-decarboxylation reaction of the β-phenyl-γ-lac- tam (S)-5a with 6N HCl was carried out, producing the (S)-Phenibut hydrochloride 6a in 85% yield. The absolute stereochemistry of 6a was confirmed by the comparison of the optical rotation data and chiral HPLC spectrum of 5a with reported data [11] [12]. Under identical conditions, the β-phenyl-γ-lactam (R)- 5a was transformed into (R)-Phenibut hydrochloride 6a in 81% yield (Scheme 2).
General procedure: The S-β-phenyl-γ-lactam 5a (96 mg, 0.32 mmol) and 6N HCl (5 mL) was refluxed for 3.5 h. After cooling, the reaction mixture was washed with DCM. The components in water phase were concentrated in vacuum under reduced pressure to afford (S)-Phenibut 6a in HCl salt form as a white solid.
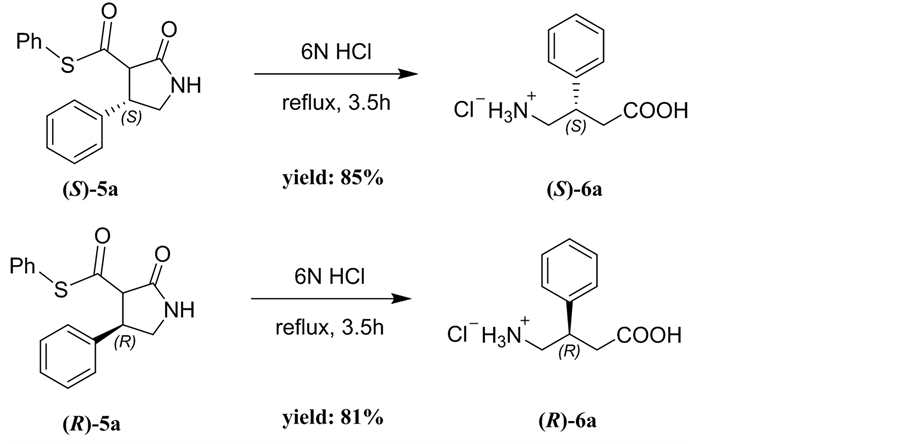
Scheme 3. Preparation of (S)/(R)-Phenibut hydrochloride 6a.
(S)-4-amino-3-phenylbutanoic acid hydrochloride (6a). White solid; yield: 85%; mp: 194˚C - 195˚C, [a]D25 = +6.1 (c = 0.25, H2O); 1H NMR, (400 MHz, CD4O) δ: 7.43 (d, 2H), 7.37 (m, 3H), 3.40 (m, 2H), 3.25(q, 1H), 2.84 (dd, J = 5.5 Hz, J = 16.1, 2H), 2.75(dd, J = 5.5, J = 9.8 Hz, 2H) ppm. IR (KBr): ν 1700, 1552, 1441, 1381, 981, 751, 657, 639 cm−1;
(R)-4-amino-3-phenylbutanoic acid hydrochloride (6a). White solid; yield: 81%; mp: 194˚C - 195˚C; [a]D25 = -5.0 (c = 0.25, H2O); 1H NMR and IR date are identical to (S)-6a.
3. Conclusion
In conclusion, we have developed the first example of cinchona alkaloid-derived thioureas-catalyzed asymmetric Michael addition of S, S’-diphenyldithiomalo- nate to trans-β-nitroolefins. An efficient asymmetric synthesis of biologically attractive chiral GABA derivatives (S) or (R)-Phenibut was readily accomplished by reduction/cyclization/ hydrolysis sequential reactions. Easily obtained substrates, catalysts and a simple experimental procedure constitute peculiar advantages of this method. Investigations aimed at developing more effective transformation of β-aryl-γ-nitroalkanes adducts to GABA derivatives without any effect on enantioselectivities are currently ongoing in our laboratory.
Acknowledgements
The financial support of National Major Scientific and Technological Special Project for “Significant New Drugs Development” (No. 2014ZX09J14104-06C) and the Young Scholar Foundation of the Fourth Military Medical University are gratefully acknowledged.