Combined Experimental and Computational Investigation of 2-(2-Hydroxyphenylimino) Phenolic Derivatives: Synthesis, Molecular Structure and NLO Studies ()
1. Introduction
Schiff bases are important class of organic compounds have long attracted attention, owing to their remarkable biological and pharmacological properties, such as antibacterial, antiviral, antineoplastic and antimalarial activities. The functional applications of the azomethine group of Schiff base derivatives enable their use in numerous fields, they have
Ø nucleophilic imine group,
Ø an imine carbon that has both electrophilic and nucleophilic character,
Ø configurations isomerism from the presence of the C=N double bond.
These structural features of the Schiff base compounds give its physical and chemical properties [1] [2] [3] . Schiff bases constitute some of the most valuable groups of biomolecules. First reported in 1864 by Hugo Schiff , these compounds gained notoriety due to the ease way of preparation from commercially available inexpensive aldehydes/ketones and primary amines. The azomethine linkage (>C=N-) allows rapid access to vast libraries of structurally diverse molecular hybrids with interesting biological properties, including antifungal, antibacterial, antimalarial, anti-inflammatory, antiviral, antioxidant, pesticidal and in-vitro/ in-vivo inhibitory effects against experimental tumor cells. The electrophilic car- bon and nucleophilic nitrogen in (>C=N-) core confer to Schiff bases the possibility to interact with several nucleophilic and electrophilic biological species, which can lead to enzymes inhibition or DNA replication impairment. Then, Schiff bases are promising as lead compounds for the rational design of novel cytotoxic and cytostatic small molecules with a mechanism of action that may differ from that of clinically approved anticancer agents [3] [4] [5] [6] .
In addition, Schiff base compounds high potential applications in non-linear optical (NLO), optical communication, optical signal processing and transmission, optical data acquisition and storage, optical computing, and especially op- tical limiting effects utilized in the protection of optical sensors and human eyes from high-intensity laser beams [7] [8] [9] [10] . In the present work, we have synthesized certain salen based Schiff base compounds, 2-(2-hydroxyphenyli- mino) phenolic derivatives viz. salcylaldehyde (SA), o-hydroxyacetophenone (AA), o-vanillin (VA) and 2-hydroxy-1-naphthaldehyde (NA), with o-aminophenol [11] [12] [13] [14] and characterized by FT-IR, UV-Vis, 13C{1H}-NMR techniques, Cyclic Voltammetry studies. Moreover, theoretical studies were carried out on the molecular structure using density functional methods (B3LYP) invoking 6-311G(d,p) basis set. The energy of the highest occupied molecular (HOMO) orbital and lowest unoccupied (LUMO) molecular orbital have been predicted.
2. Experimental
Materials and Methods
All the chemicals and solvents used were purified and dried by standard methods. FT-IR spectra were recorded as KBr pellets with a PerkinElmer FT-IR spectrometer in 4000 - 400 cm−1 range. Microanalyses were carried out with a Vario El AMX-400 elemental analyzer at STIC, Cochin University of Science and Technology, Kerala, India. Electronic spectra were recorded in CH3CN as solvent with an Ocean optics spectrophotometer USB 4000. NMR spectra were also recorded on Bruker AVANCE III 500 MHz (AV 500) spectrometer; chemical shifts are expressed in ppm (δ units) relative to TMS signal as internal reference in DMSO.d6.
The Schiff base compounds (Scheme 1) were prepared by the reported literature procedure [11] [12] [13] [14] with modification of the substitutions and the purity of the Schiff bases were checked by TLC. Melting points were recorded with a Inlab, India micro heating table and were uncorrected.
To an ethanolic solution of salcylaldehyde (SA), o-hydroxyacetophenone (AA), o-vanillin (VA) and 2-hydroxy-1-naphthaldehyde (NA) (2 cm3, 20 mmol), with o-aminophenol (2 cm3, 20 mmol) were added with stirring. The mixture was then refluxed for 4 - 6 hours with the controlled conditions about 140˚C - 180˚C. On cooling the solution, a solid compound which separated out was filtered, dried and recrystallized from ethanol/DMSO (75:25). The purity of the ligand was checked by TLC. Yield, 85%, m.p. 143.6˚C - 245˚C.
All the salen compunds were stable at room temperature, non-hygroscopic and insoluble in water partially soluble in methanol, ethanol and soluble in CH2Cl2, CHCl3, DMF, DMSO, CH3CN, etc.
3. Computational Methods
All the computational studies have been carried out with the GAUSSIAN 03W program package [15] . Density functional theory (DFT) method has been applied because of its excellent compromise between computational time and description of electronic correlation and qualitative structure-activity relationships (QSAR) studies [16] . B3LYP, a hybrid functional of the DFT method, which consists of the Becke’s three parameters exact exchange functional B3 [17] combined with the non-local gradient corrected correlation functional of Lee-Yang- Parr (LYP) [18] has been used. The standard triple split valence basis set 6-311G [19] with a set of d, p polarization functions on heavy atoms and hydrogen atoms are used throughout the computational process.
A fully relaxed potential energy scan was carried out against the dihedral angle C2-C1-C7-N1 (Scheme 2) at B3LYP/6-311G(d,p) level, the minimum energy conformations from the energy scan, a further geometry optimization was performed at the same level of theory. Vibrational frequencies of the optimized structures were computed using the same level of theory and thermodynamic
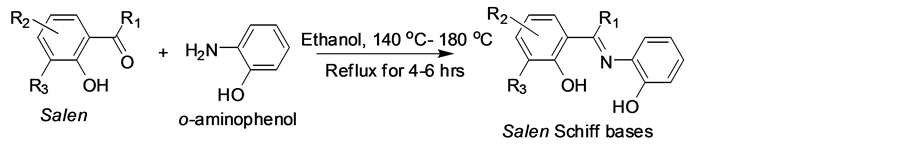
Scheme 1. Preparation of Schiff base compounds.
Scheme 2. Potential energy scan (PES) for salen compounds.
corrections [20] [21] were obtained at 298 K and 1 atm, and added to electronic energies.
Calculated electronic properties such as dipole moment, first static hyper-po- larizability HOMO and LUMO energies, MEP, energy gap, electronic affinity (EA), electronegativity (χ), hardness (η), softness (S), electrophilic index (w) and ionization potential (IP) have been studied for all the four compounds under consideration. The DFT-based reactivity descriptors were obtained from the Equations (1)-(4) [22] [23] [24] [25] which play an important role in many areas of research.
Electronegativity (χ)
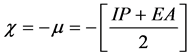 (1)
(1)
Hardness (η)
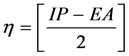 (2)
(2)
Softness (S)
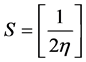 (3)
(3)
The experimental polarizability is obtained as an average polarizability, given
by 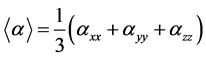 and the computed average polarizability is also
and the computed average polarizability is also
presented.
4. Results and Discussion
4.1. Experimental Results
4.1.1. Elemental Analysis and FT-IR Spectral Data
The elemental analysis data (Table 1) for the all Schiff base compounds are well agreed with the proposed molecular formulae.
The FT-IR spectroscopy is a powerful tool for the assignments of fundamental functional group determinations of organic compounds. In the title compounds frequencies of the functional groups viz., ν>C=N-, νph-OH, νph-C-O, νph-N=C- and ν-N=CH-/ν-N=C(CH3)- are of great importance in the infrared spectra. The infrared spectra of all the four Schiff bases (Figure 1) exhibited a strong band in the 1692 - 1642 cm−1 region due to the characteristic of azomethine n(C=N) group. All the title compounds were displayed a band in the region 3399 - 3308 cm−1, which could be due to ν(ph-OH). A strong band observed at 1373 - 1362 cm−1 in
![]()
Table 1. Lemental analysis of Salen compounds.
the four compounds has been assigned to phenolic C-O stretching and a band due to ph-N=C- of aminophenol group exhibited in the region 1274 - 1296 cm−1. Further the spectra of all the Schiff bases showed a weak band in the 3020-3069 cm−1 region due to -N=CH-/-N=C(CH3)- groups of NA, SA, VA and AA [14] .
Theoretically all the fundamental vibrations were active in IR. The results showed that the DFT (B3LYP) method applied in this work leads to vibrational wavenumbers which are in good agreements with the experimental data. It is noteworthy that the very important role of vibrational frequencies salen compounds, based on experimental data as well as theoretical calculations, the computed vibrational frequencies in Table 2 and Figure 1 for SA, AA, VA and NA at the DFT level of theory are in acceptable agreement with the experimental data.
4.1.2. UV-Vis Spectra
The UV-Visible spectra of the salen compounds (Figure 2) exhibit mainly two bands. The first band on the higher energy side, the range lmax= 350 - 385 nm, due to the excitation of the p-electrons (p®p* transitions) of the aromatic azo-
![]()
Table 2. Vibrational frequencies of salen compounds (Experimental & Theoretical).
methine group. The second band, lmax = 450 - 480 nm range is assigned to an intramolecular charge transfer (ICT transition) involving the salen compounds. This band observed in salicylaldimine compounds of salen derivatives is facilitated by the presence of intramolecular hydrogen bonding between the -OH group and the azomethine nitrogen [26] .
4.1.3. 13C{1H}-NMR Spectra
The formations of Schiff base were conveniently monitored by peak ratios in the 1H-NMR spectra. 1H-NMR spectra of all the four compounds (Table 3 and Figure 3) were taken in DMSO.d6 solvent. The aromatic region was a set of multiplets in the range 6.6 - 8.4 ppm for all the Schiff bases, while the azomethine proton of SA, VA and NA compounds were observed in the range 9.0 - 9.5 ppm. The phenolic-OH protons of all the four Schiff bases were observed as a singlet in the region 13.8-15.6 ppm. 1H-NMR spectra exhibited a strong peak at 3.8 and 3.5 ppm for -OCH3 protons of VA and -CH3 protons of AA salen compounds.
13C-NMR spectral data (Table 3 and Figure 3) were consistent with 1H-NMR spectral data. The methyl carbon of aliphatic substituents, azomethine (>C=N) for all the Schiff bases and for Ph-C-CH3 of AA and Ph-OCH3 of VA compounds were 45 and 56 ppm respectively. The resonance observed at 108 - 178 ppm was assigned to the phenyl group carbon of salen compounds.
4.2. DFT Study
4.2.1. Conformational Analysis and Optimized Geometries
At the minimum energy conformations (Figure 4) obtained from the energy scan, further geometry optimization was performed with the B3LYP/6-311G (d,p) basis set. No geometrical parameter constraint was imposed during the optimization, except those favoring the stabilizing effects due to hydrogen bonding between two adjacent -OH groups. The optimized structures of the most stable conformers of neutral form of salen compounds are shown in Figure 5.
The optimized geometrical parameters are shown in Table 4 (Supplement materials). From the data of bond distances and bond angles, it can be seen that no significant geometrical change has been observed for all the compounds. From the data of dihedral angle in Table 4, it can also be seen that compounds with o-hydroxyl group in A-ring are completely planar, while others have some
![]()
Figure 4. PES scan optimization of salen compound AA.
![]()
Table 3. NMR Chemical Shifting of Salen compounds.
degree of deviation from the planarity due to the torsion between A-ring and the plane of aminophenol system B-ring (Scheme 2).
In Table 5 the dipole moment of four compounds at B3LYP/6-311G(d,p) level of computation have been reported. The Dipole moment values lie in the range of 1.7675D to 4.8823D for salen compounds. These values are rather high, reflecting the numerous polarized hydroxyl or carbonyl functions distributed over the structures. The three structures of salen compounds differ only by the presence of methyl at C7, methoxy at C3 and naphthyl groups in the A-ring and there is a little difference in the dipole moment value. The molecular dipole moment is used to measure bond polarities and charge densities in a molecule.
![]()
Table 5. Electronic energies for Schiff base neutral molecule and radicals in gas phase at B3LYP/6-311G(d,p) level of theory.
Hardness,![]() .
.
Bond polarity is one of the factors that determine the physicochemical property of molecules. The calculated values of the total dipole moments, which signifies the relatively polarized nature of the systems and they are soluble in polar solvents like CH3CN, DMSO, CHCl3, etc.
The computed NMR chemical shifts for SA, AA, VA and NA at the DFT level of theory are in acceptable agreement with the experimental data. Differences between the calculated and measured values may be a result of solvent interactions.
The 13C-NMR chemical shifts of selected carbons were calculated on the optimized structures of SA, AA, VA and NA using GIAO/DFT method with B3LYP/6-311(d,p) basis set for all atoms. Calculated and measured 13C chemical shifts of selected atoms are numbered in Figure 6.
4.2.2. HOMO-LUMO Orbital Distribution
4.2.3. Spin Density Distribution
The spin density is often considered to be a more realistic parameter which provides a better representation of the reactivity [6] [9] of salen moiety. It should be pointed out that the more delocalized the spin density in the salen compound is, the easier the radical is formed [5] . In the Figure 8, the spin densities of all the radicals mainly distribute on the phenolic oxygen atom and the phenyl ring A
![]()
Table 6. Total energies, Frontier orbital energies, Softness, Egap and EI in gas phase calculated at B3LYP/6-311G(d,p) level of theory.
![]()
![]()
Table 7. All β(a.u.) components and β(tot) × 10−31 (esu) value calculated using DFT level of theory for salencompounds (1 a.u. = 8.6393 × 10−33esu).
and B. Comparing all the radicals, we find that the radicals have almost the same spin density distribution with each other, indicating the presence of additional hydroxyl or o,o-dihydroxyl group on A and B-ring has almost no influence on the spin density distribution [27] [28] .
4.2.4. Molecular Electrostatic Potential Surface (MEP) Study
Electrostatic potential surfaces are mainly used to study the reactive species of electrophilic or nucleophilic attacks/substitution in the chemical reactions, biological process, catalysis and also molecular modeling. Electrostatic potential mapped surface displays the molecular size, shape and potential values. In this study, 3 dimensional surfaces of mapped onto the constant electron density surface is as shown in Figure 9. Different values of electrostatic potential at the surface are represented by different colors [29] .
4.2.5. Computed Non-Linear Optical (NLO) Properties
The NLO response calculation was performed on the optimized geometry using the same level of theory. The first static hyperpolarizability is a third rank tensor that can be described by a 3 × 3 × 3 matrix. The 27 components of the 3D matrix
![]()
Figure 9. Molecular Electrostatic Potential surface diagram of AA.
can be reduced to 10 components due to the Kleinman symmetry [27] [30] (βxyy = βyxy = βyyx, βyyz = βyzy = βzyy, ..., likewise other permutations also take same value). It can be given in the lower tetrahedral format. The output from Gaussian 03W provides 10 components of this matrix as βxxx, βxxy, βxyy, βyyy, βxxz, βxyz, βyyz, βxzz, βyzz and βzzz respectively. Many types of hyperpolarizabilities have been discussed in the literature [31] . When reporting a single value of β, one of the common formats is to simply treat the three independent values for β as a quasi-Pythagorean problem and solve for the average β by Equation (5):
![]() (5)
(5)
The complete equation for calculating the magnitude of the total first statichyperpolarizability from Gaussian 03W output is given as Equation (6):
![]() (6)
(6)
Since these β values of the first static hyperpolarizability (β) tensors of the output file of Gaussian 03W are reported in atomic units (a.u.), the calculated values were converted into electrostatic units (1 a.u. = 8.6393 × 10−33 esu). The first static hyperpolarizability (β) values of these Schiff bases were calculated under static electronic field by the DFT (B3LYP/(6-311G(d,p)) method. From Table 7, compounds NA, VA and AA have larger the first static hyperpolarizability (βtot) values than those of compound SA. As mentioned above, the DFT results show that the LUMO orbitals of all compounds were obtained from the linear combination of the orbitals of phenyl moiety, the compounds AA, VA and NA with an electron-donating and withdrawing group (CH3, OCH3 and naphthyl) have a larger βtot value compare with compound SA. Hence the compound VA was predicted to have larger NLO property. The energy difference (∆Egap) between the HOMO and the LUMO orbitals 3.5506, 4.3538, 3.8368, 3.4014 eV for the compounds SA, AA, VA, NA respectively has a larger influence on the βtot value [32] [33] . To understand the relationship between the static hyperpolarizabilities and the HOMO-LUMO energy gap, viz. the compound VA (3.4014 eV) lower the HOMO-LUMO energy gap show that the larger the βtot (13.455 a.u) value.
4.3. Conclusions
Schiff bases (1-4) have been successfully synthesized and characterized by elemental analysis, FT-IR, UV-Vis, NMR spectroscopy and cyclic voltammetry. The evaluation of the quantum mechanical studies reveal significant activity in structure optimization, PES, vibrational study, NMR Chemical shifts, spin density, hardness, electronegativity, dipole moment, EHOMO/ELUMO, ∆Egap and Softness which provide the evidence for a very strong positive correlation between experimental and theoretical predictions. These compounds also have towards considerable antioxidant activity along with potential to prevent DNA oxidative damage by free radicals.
Comparison between the four considered molecules indicates compound VA that requires the lowest energy for both H atom and electron transfer mechanisms. This theoretical approach confirms the important role of A and B ring in exhibiting antioxidant properties. Inspection of deprotonation processes of dihydroxyl groups have shown that, π electron delocalization of phenyl ring A and B (evidenced from UV-Vis study) plays a major role in the stabilization of products and thus in the lowering of the associated energies. The variables related to the chemical potential allow classifying salen type of Schiff base compounds that has the tendency to give electrons more than to attract them, which demonstrates biological importance.
In addition to that in order to understand the relationship between the βtot values and the substitution groups of salen compounds, the frontier orbital compositions have been analyzed and the energy gaps between the HOMO and LUMO orbitals were also calculated. The compound VA with electron-donating group (-OCH3) will produce the larger βtot value (13.455 a.u) than lower βtot (10.003, 9.244, 4.359 a.u) for the compounds NA, AA, SA respectively. The energy gaps between the HOMO and LUMO orbitals show that the lower the HOMO-LUMO energy gap, larger the first static hyperpolarizability (βtot).
The findings of this work sustenance the view that some of these synthesized compounds are promising sources of potential drugs that may be efficient as preventive agent(s) in some diseases.