Microwave-Assisted Facile and Rapid Esterification of Amino Acids I: Esterification of L-Leucine from Batch to Flow Processes and Scale-Up ()
1. Introduction
Microwaves are electromagnetic waves with frequencies ranging between 300 MHz and 30 GHz. They are often employed in radars, wireless communication devices, and as heating tools in the form of microwave ovens [ 1 ]. In recent years, microwave heating has also been widely used in laboratories as a high throughput tool for preparative chemistry [ 2 - 7 ]. Indeed, a number of reports have been published discussing the application of microwave irradiation in organic synthesis for accelerating reaction rates and improving product selectivity. However, to date, the majority of studies on microwave-assisted synthesis have been carried out using desktop-scale reactors, despite various reports on the scale-up of microwave syntheses for potential application on an industrial scale [ 8 - 11 ].
In the context of organic synthesis, amino acid esters are widely employed as inter-mediates for a wide range of applications, such as medicinal chemistry [ 12 ], peptide synthesis [ 13 ], asymmetric synthesis [ 14 ], dye-sensitized solar cells [ 15 ], and polymer chemistry [ 16 ]. To date, the principal method of amino acid ester synthesis using alcohols has been the Fischer-Speier esterification procedure [ 17 ], in which an amino acid and an alcohol are dehydrated in the presence of an acid catalyst, such as hydrogen chloride [ 18 ], sulfuric acid [ 19 ], polyphosphoric acid [ 20 ], p-toluenesulfonic acid [ 21 , 22 ], ion-exchange resins (e.g., Amberlyst® 15) [ 23 ], or trimethylchlorosilane [ 24 ]. However, as the amino group present in amino acids can form an amide moiety through bonding with the carboxylic group of another molecule, the amino group must be protected prior to esterification. This is typically carried out using an equimolar amount of acid to produce the species shown in Eq. (1). Alternatively, an N-protected Boc-amino acid can be employed [ 25 ], which is prepared from the Bocanhydride rather than an acid. In terms of the esterification reaction itself, amino acids can be esterified using a combination of methanol and thionyl chloride, where the acid chloride reacts with methanol to form the ester [ 26 ]. This reaction proceeds smoothly at relatively low temperatures (i.e., ~50˚C); however, it requires an excess of the toxic thionyl chloride reagent to drive the reaction to completion. As such, safer and more efficient methods of amino acid esterification are required for application in industry.
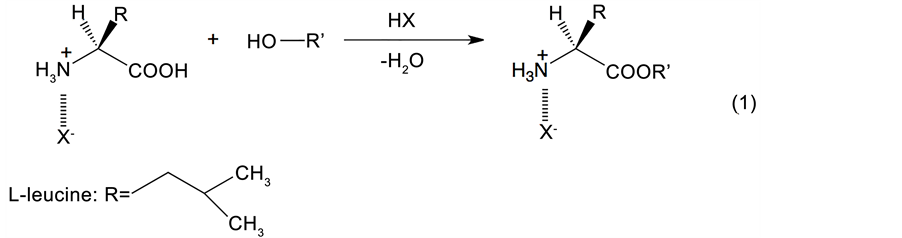
As mentioned above, the Fischer-Speier esterification is the most common method for amino acid ester synthesis using alcohols. However, this process generally requires large quantities of hazardous reagents, is time consuming, and forms various hazardous by-products, which must subsequently be removed via cumbersome post-processing methods. For example, during this reaction, a large volume of toluene is required to produce an azeotrope with water. However, toluene is particularly toxic, and its release into the atmosphere is the largest among the pollutant release and transfer register (PRTR) chemicals of a number of countries. Therefore, a new synthetic method that does not require the use of toluene as a solvent is of particular importance for industrial applications.
Due to the high polarity and the ionic character of amino acids (see Eq. (1)), micro-wave heating is expected to be a favored technique for their esterification reactions. In-deed, the esterification of N-protected amino acids via microwave synthesis has been reported in recent years [ 27 - 29 ]. Moreover, Martinez-Palou’s group reported the esterification of unprotected amino acids with long-chain alcohols using a small-scale micro-wave reactor in the context of combinatorial chemistry [ 30 ]. However, the origins of microwave effects on the amino acid esterification process have not yet been clarified, and to the best of our knowledge, the scale-up of this reaction has also not been described yet.
Thus, we herein report our studies into the effects of microwave irradiation on the esterification of amino acids with ethanol and n-butanol, using L-leucine as a representative amino acid. n-butanol was selected as both a solvent and a reagent for a number of reasons. Firstly, the butyl ester of L-leucine is widely utilized as an intermediate in the preparation of pharmaceutical products, such as valsartan, which is employed in the treatment of high blood pressure and congestive heart failure [ 31 ]. In addition, n-butanol can form an azeotrope with water, whose boiling point (117.7˚C) is close to that of toluene (i.e., 110.6˚C). Following optimization of the reaction with n-butanol, the batch and flow processes of this reaction will be compared, with the overall aim of scaling-up the process for industrial-scale application.
2. Materials and Methods
Here, we first compared the efficiencies of microwave and conventional oil bath heating processes in amino esterification of L-leucine using batch reactors. Then two types of microwave flow reactions were examined using a desktop-scale tubular reactor and a semi-bench-scale tubular reactor in this reaction.
2.1. Materials
All reagents were purchased from Wako Pure Chemical Ind., Ltd. (Japan). L-Leucine (Leu, ≥99%) and p-toluene sulfonic acid (PTS, ≥99%) were used with-out further purification. Ethanol (EtOH) (≥95%) and n-butanol (BuOH, ≥99%) were distilled prior to use.
2.2. Analysis of the Reaction Products
The yield and the selectivity of the L-leucine esterification reaction were determined using 1H NMR spectroscopy (Avance III 400M NMR spectrometer, Bruker, Germany). All spectra were recorded at 400 MHz. Deuterated chloroform (CDCl3, Wako Pure Chemical Ind., Ltd. Japan) and tetramethylsilane (TMS, Wako Pure Chemical Ind., Ltd., Japan) were used as the solvent and internal standard for all NMR measurements, respectively.
2.3. Measurement of Complex Dielectric Parameters
The dielectric properties of the various reagents were measured via a coaxial probe method using an open-ended coaxial probe, as described elsewhere [ 32 ]. The measurement system consisted of a custom-built separable flask, a high-temperature dielectric probe (85070E, Agilent Technologies, USA) operating between −40 and 200˚C, and a vector network analyzer (8720ES, Agilent Technologies) operating between 0.2 and 20 GHz. A PCMCIA-GPIB interface (National Instruments, USA) was employed to connect the vector network analyzer to a PC equipped with data-collecting software (85,070, Agilent Technologies) capable of calculating complex dielectric properties using reflected signals (S11) from the vector network analyzer. The flask was heated from the bottom and stirred using a stirring hotplate. Measurements were carried out at various temperature intervals after stabilization for 1 min.
The system was calibrated prior to each measurement to compensate for small variations in the cable position, the connections, the ambient temperature, and other factors capable of affecting system performance. The testing probe was calibrated using a short circuit (a gold-plated precision shorting block included in the 85070E probe kit), an open circuit (air), and a known load (deionized water, G18.2 MΩ, Milli-Q, Millipore, at 25˚C).
2.4. Reaction Apparatus
2.4.1. Batch-Type Microwave Reactor
The batch-type microwave-assisted esterification of L-leucine was carried out using a multi-mode microwave apparatus (Reactor model SMW-087, 2.45 GHz, maximum power output 770 W, Shikoku Instrumentation Co., Ltd., Japan) equipped with a magnetic stirrer and three metal pipes (41 mm f × 101 mm length) to act as a reflux condenser and a thermometer.
A mixture of L-leucine, alcohol, and acid catalyst were placed into a glass round-bottomed flask equipped with a reflux condenser, a magnetic stirrer, and a fiber optic thermometer (AMOTH FL-2000, Anritsu Meter Co., Ltd., Japan). The flask was then placed inside the cavity of the microwave reactor, and was subjected to microwave irradiation at the reflux temperature. The reaction temperature was controlled automat-ically using the fiber optic thermometer, and all reactions were carried out under atmospheric pressure.
2.4.2. Desktop-Scale Microwave Tubular Reactor
The experimental setup employed for carrying out the flow reaction using a desk-top-scale microwave tubular reactor is shown in Figure 1. A glass tube (40 mm × 300 mm) was placed horizontally through the microwave reactor, which was the same reactor employed for the batch reaction. One end of the glass tube was connected to a rotating mortar (SE-1000, Tokyo Rikakikai Co., Ltd., Japan), while the other end was connected to a reflux condenser and a storage tank. A Teflon baffle was placed in the tube to enhance mixing of the reactant, and the solution containing the reactants was fed into the tube using a peristaltic tube pump (EYELA MP-1000, Tokyo Rikakikai Co., Ltd., Japan). The flow rate was controlled using a second peristaltic tube pump to maintain a depth of ~13 mm in the reactor. Finally, the reaction temperature was measured using the previously described fiber optic thermometer.
2.4.3. Semi-Bench-Scale Microwave Tubular Reactor
Figure 2 shows the experimental setup of the semi-bench-scale microwave tubular reactor. In this case, the reaction was carried out inside a Teflon tube (25.4 mm × 950 mm) placed vertically inside a cylindrical cavity composed of SUS 304 stainless steel (Fuji Electronic Industrial Co., Ltd., Japan, 160 mm × 950 mm) with a microwave inlet window. The cavity was equipped with a microwave generation device (SMW-101, 2.45 GHz, maximum output power 1500 W, Shikoku Instrumentation Co., Ltd., Japan), a pressure gauge, a pressure regulator, and a safety valve. The raw material was originally stored in a feed tank prior to its supply to the bottom of the cavity using a helical pump (PC Pump 3NY06, Heishin Ltd., Japan) and subsequent heating by microwave irradiation. The raw material was then introduced into the pipeline at a flow rate of 20 mL·min−1, and the temperature was set at either 120˚C or 140˚C. The reaction temperature was controlled automatically by means a K-type thermocouple located at the center of the reaction tube. After heating, the product was stored in a storage tank.
2.5. Esterification of L-Leucin
The typical reaction procedures for the various setups were as follows.
2.5.1. Microwave-Assisted Batch Reaction
A mixture of L-leucine, n-butanol, and p-toluene sulfonic acid (Leu/BuOH/PTS = 1.0/33.0/1.2 molar
![]()
Figure 1. The flow type desktop-scale microwave reactor.
![]()
Figure 2. Semi-bench-scale microwave tubular flow reactor. (a) Schematic experimental setup of the reactor, and (b) photo of the reactor.
ratio, 95 mL) was added to a 100-mL round-bottomed flask, which was subsequently placed in the cavity of the microwave reactor. The mixture was then subjected to microwave irradiation, with the output power controlled automatically to maintain the reflux temperature. After completion of the reaction, the resulting solution was analyzed by 1H NMR spectroscopy.
2.5.2. Conventionally-Heated Batch Reaction
A conventionally-heated batch reaction was carried out using an oil bath (model no. BO500, 770 W, Yamato Scientific Co., Ltd., Japan). The remainder of the experimental setup was as previously described for the batch microwave process. After placing the reaction mixture (Leu/BuOH/PTS = 1.0/33.0/1.2 molar ratio, 95 mL) into a 100-mL flask, the flask was dipped into an oil bath preheated to 20˚C higher than the reaction temperature to ensure that the desired temperature was achieved inside the reaction flask. The temperature of the reaction gradually increased from 25˚C to reflux over 6 min, after which time the target temperature was maintained for 30 min under atmospheric pressure. After completion of the reaction, the resulting solution was analyzed by 1H NMR spectroscopy.
2.5.3. Desktop-Scale Microwave-Assisted Flow Reaction
A small-scale flow reaction was carried out using a desktop-scale microwave tubular reactor. Prior to the reaction, the reaction tube was half-filled with the reactant mixture described above, and the glass tube was rotated at 10 rpm. This rotation speed was maintained constant throughout the reaction. After commencing rotation, the reactant mixture stored in the feed tank was pumped into the tube at the desired flow rate (0.25 - 10 mL·min−1) via the reactor inlet, then heated to reflux under microwave irradiation over 60 min. The effluent containing the product mixture was then collected in the storage tank and was analyzed by 1H NMR spectroscopy.
2.5.4. Microwave-Assisted Semi-Bench-Scale Flow Reaction
The same reactant mixture employed for the batch reaction was pumped from the storage tank to the reaction tube at a flow rate of 20 mL·min−1 and the reaction temperature was automatically controlled at either 120˚C or 140˚C, while the pressure was maintained below 0.5 MPa. Approximately 600 mL of reactant was required to fill the reactor. The effluent containing the product mixture was then collected in the storage tank and analyzed by 1H NMR spectroscopy.
3. Results and Discussion
3.1. Esterification of Amino Acids with N-Butanol or Ethanol in a Batch System
Table 1 shows a comparison of the results obtained for the esterification reactions of L-leucine with n-butanol and ethanol under microwave heating and conventional heating. As previously mentioned, for conventional heating, the temperature of the oil bath was set 20˚C higher than the boiling temperature of the alcohol employed. After heating gradually to reflux over 6 min under atmospheric pressure, the reaction temperature was maintained for 30 min to give the desired butylated ester in 63.7% yield and the desired ethyl ester in 3.9% yield. This lower yield for the ethyl ester was likely due to the lower temperature of the ethanol system (ethanol boiling point = 78.4˚C). In addition, as ethanol is more miscible with water than butanol, this can inhibit the removal of the water by-product from the reaction solution and lower the ester yield.
In contrast, significantly higher yields were obtained under microwave irradiation: L-leucine butyl ester and L-leucine ethyl ester were obtained in 100% and 35.7% yields, respectively, after a reaction time of 10 min. These enhanced yields were likely due to reflux being achieved after only 2 min, thus allowing a shorter reaction time and reduced side-product formation. The lower yield for the ethyl ester can be accounted for as described previously.
To investigate the differences in heating rates between the two methods more closely, the variation in reaction temperature with time for the esterification of L-leucine with n-butanol was examined, as outlined in Figure 3. Under microwave heating, the temperature of the reaction mixture rapidly increased to 127˚C after 2 min, prior to dropping slightly to 124˚C for the remainder of the reaction. In contrast, under oil bath heating, the oil bath was heated to 140˚C prior to the reaction, and then the reaction flask was dipped into the bath and the internal (reaction) temperature recorded. As shown, the temperature of the reaction solution increased gradually, reaching the reflux temperature of 117˚C after 6 min. The dotted line shown in the figure indicates the differences in temperature between microwave heating and conventional heating at each point during the reaction. As indicated, microwave heating led to higher temperatures than oil bath heating until the systems reached ~60˚C, after which point a difference of 7˚C - 10˚C remained apparent even under reflux conditions. This phenomenon is known as super-heating, and it relates to the temperature of a solution under microwave irradiation being higher than achievable under conventional heating methods even under reflux conditions.
To date, a number of examples of superheating have been reported in the literature [ 33 ] [ 34 ]. For example, Baghurst and Mingos proposed a mechanism for superheating in which under conventional
![]()
Table 1. Comparison of microwave heating and oil bath heating in the esterification of L-leucine with n-butanol or ethanol. Reaction conditions: Leu/(BuOH or EtOH)/PTS = 1.0/(33.0 or 52.3)/1.2 mole ratio, heated for 10 min.
aMW: microwave heating, CH: conventional heating by an oil bath.
heating, the reflux of a liquid is caused by the superheating of bubbles emerging from a vapor embryo trapped inside the crevice of the vessel wall, where the temperature surrounding the vessel wall is higher than that of the center of the liquid body. In contrast, under microwave heating, the opposite temperature distribution is observed [ 35 , 36 ]. As such, when the temperature surrounding the vessel wall reaches the reflux temperature, the temperature of the liquid body is lower than that surrounding the vessel wall under conventional heating, while it is higher under microwave heating.
As determined from Figure 3, the initial rates of temperature increase in the micro-wave and conventional systems were 166˚C min−1 and 27˚C min−1, respectively. This indicates that microwave irradiation transfers heating energy to the solution at a faster rate than in conventional heating, and so this could also account for the acceleration effects observed under microwave heating. As such, the mechanism of superheating and the high rate of energy transfer appear to account for the accelerated reactions observed herein upon the use of microwave heating.
3.2. Measurement of the Dielectric Properties of Reactants under the Reaction Conditions
As microwaves directly heat dielectric materials, the efficiency of this method depends on the dielectric properties of the material being heated. The dielectric property of a material can be expressed using a complex dielectric constant,
, which is composed of the dielectric constant,
, and the dielectric loss factor,
, to give
, where j is an imaginary number. The power dissipated into or absorbed by the material by the conversion of electromagnetic energy into heat can be expressed by Eq. (2), where ω is the angular frequency, ε0 is the dielectric constant of free space (ε0 = 8.854 × 10−12 F·m−1),
is the dielectric loss factor, E is the electric field strength, and V is the volume of the material.
(2)
![]()
Figure 3. Reaction time course of the esterification of L-leucine with n-butanol. Circle: microwave irradiation, rectangle: oil bath heating, dashed line: difference of the temperatures between microwave irradiation and oil bath heating.
The dielectric constant,
, indicates the ability of a material to store electromagnetic energy through polarization. In contrast, the dielectric loss factor,
, is an indicator of the ability of a material to convert the stored energy to heat. In this context, we investigated the dielectric properties of both the reaction mixture (Leu/BuOH/PTS = 1.0/33.0/1.2 molar ratio) and of n-butanol as a function of temperature between 20˚C and 100˚C as shown in Figure 4. It should be noted that the measurement of the properties of n-butanol under reflux was hampered by the formation of bubbles around the sensor. However, we found that its dielectric constant increased gradually from 4.3 to 9.2 with an increase in temperature from 30˚C to 100˚C. The dielectric constant of the mixture also increased from 4.7 (25˚C) to 9.3 (100˚C), giving higher values than n-butanol over the temperature range examined herein (Figure 4(a)).
![]() (a) (b)
(a) (b) ![]() (c) (d)
(c) (d)
Figure 4. Dielectric properties of a mixture of reactants (Leu/BuOH/PTS =1.0/33.0/1.2 molar ratio, squares, solid lines) and n-butanol (circles, dashed lines). (a) Dielectric constants, (b) dielectric loss factors, (c) tangent loss factors, and (d) half penetration depths.
In addition, the dielectric loss factors of both the mixture and of n-butanol alone initially increased upon increasing the temperature, although a subsequent decrease was observed beyond the maximum values at 70˚C - 80˚C. As for the dielectric constant, the values of the dielectric loss factors were also significantly higher for the mixture than for n-butanol, as shown clearly in Figure 4(b). This higher dielectric loss factor for the L-leucine mixture compared to n-butanol at ~100˚C could therefore account for the enhanced esterification activity under microwave heating compared to conventional oil bath heating.
The penetration depth, d (at half power) of microwave energy is also a key factor in predicting efficient heating. This factor is given by Eq. (3), where tan #Math_17# is the tangent loss factor, and λ = c/f is the wavelength of the microwave.
(3)
For our system, the power penetration depths of n-butanol and of the mixture were 14.0 and 9.6 mm, respectively at 100˚C. These penetration depths are significantly shorter than the diameter of the reaction vessel (i.e., 40 mm), which indicates that the majority of microwave irradiation is consumed close to the solution surface, and so any reactants present close to the center of the solution body will be heated indirectly by conduction from molecules closer to the surface.
3.3. Flow Reaction Process for the Esterification of L-Leucine: Small-Scale Flow Reaction
The short microwave penetration depths of the reaction solutions determined in section 3.2 suggest that from the viewpoint of energy transfer, a thinner reaction vessel is favorable for this microwave-assisted esterification. The use of a thin tube reactor was there-fore examined to clarify the efficiency of microwave heating in this reaction.
We initially investigated a stopped-flow reaction, where the reaction solution was placed in the reaction tube, then heated to reflux under microwave irradiation over 60 min. The tube containing the solution of 13 mm depth was rotated throughout the duration of the reaction. The same mixture was also placed in a 100-mL round-bottomed flask and heated at reflux under microwave irradiation. As shown in Figure 5(a), under microwave irradiation at the same power output, a 14% higher yield of the ester was obtained from the tubular reactor compared to the round bottom flask. This could be accounted for by the above-described microwave penetration depth effect.
Furthermore, Figure 5(b) shows the reaction profiles obtained for the tubular reaction under a range of flow rates. When the flow was stopped, the ester yield reached 100% after 60 min following a linear increase over the initial 30 min. Upon increasing the flow rate from 0 to 10 mL·min−1, the ester production rate decreased from 2.98% min−1 at the stopped-flow to 1.64 and 1.91% min−1 at flow rates of 0.25 and 10 mL·min−1. Indeed, under all flow rates examined herein, a linear increase in ester yield was observed up to 30 min, after which a gradual increase took place until the maximum yield for each condition was reached. Interestingly, a maximum ester yield of only 60% was obtained after 60 min at a flow rate of 10 mL·min−1 (c.f., 100% for the stopped-flow reaction).
3.4. Semi-Bench-Scale Microwave-Assisted Flow Reaction
We then examined the semi-bench-scale microwave-assisted flow reaction to determine the potential for scaling up our reaction system. After adding the various reagents to the reactor (inner volume, ~600 mL), a flow of 20 mL·min−1 was provided using the helical pump and the reaction mixture was subjected to microwave irradiation. Initially, the microwave power was automatically maintained at a lower value to control the reaction; however, after the reaction temperature reached the desired value, the microwave power was increased and maintained at the output power of 700 W. The temperature and power profiles of the reaction carried out at 140˚C are illustrated in Figure 6(a). After the temperature reached the desired value, the microwave output power automatically decreased gradually from 700 W to ~400 W, and the maximum pressure in the reactor did not exceed 0.5 MPa.
Samples of the reaction mixture were taken periodically during the reaction and analyzed by 1H NMR spectroscopy to examine the course of the reaction.
![]() (a) (b)
(a) (b)
Figure 5. Esterification of L-leucine with n-butanol using a desktop scale tubular reactor and a round-bottomed flask. (a) Comparison of the reaction vessels (circles: stopped-flow tubular reactor, squares: round-bottomed flask), and (b) comparison of the effect of flow rate on ester yield in the tubular reactor.
![]() (a) (b)
(a) (b)
Figure 6. Esterification of L-leucine with n-butanol using a semi-bench scale microwave tubular reactor. (a) Reaction profiles, and (b) variation in ester yield with time.
The variation in ester yields with time at 120 and 140˚C are shown in Figure 6(b). As expected, significant differences were observed between the two systems, as at 120˚C, the ester yield increased to reach a maximum of ~40% after 10 min, while at 140˚C, a maximum yield of 72% was achieved after 30 min. For comparison, at 140˚C, an ester yield of ~53% was obtained after only 10 min.
Under these conditions, 1.2 kg of the reactant mixture could be treated in 1 h, thus confirming the potential of this method for application in a larger scale reaction process for industrial use. In addition, to further enhance the output on a larger scale, we propose that a higher ester yield could be obtained at higher reaction temperatures by increasing the microwave power. Moreover, reagent recycling and longer reaction times may also be effective in producing higher yields.
4. Conclusion
We herein discussed our studies into the application of microwave irradiation to the solvent-free esterification of L-leucine with alcohols, namely ethanol and n-butanol. We found that under microwave irradiation, the reflux temperature of the reaction solution was higher, the reaction was accelerated, and the yield of the desired amino acid ester was increased compared with conventional heating methods. These effects are expected to be due to the more rapid transfer of energy to the reactant under microwave heating.
We also examined the dielectric properties of the reactants close to the reaction temperature, and found that the accelerating effect of microwave irradiation was caused by the higher dielectric loss factor,
, of the mixture of the amino acids, alcohol, and acid catalyst (p-toluene sulfonic acid) compared to that of n-butanol alone. In addition, the half power penetration depths of the reaction mixture and n-butanol were 9.6 - 12.6 and 14.0, respectively. This result suggested that a thinner reaction vessel was more favorable for this reaction, and this was confirmed by carrying out the esterification reaction in a tube reactor with a reactant depth of only 13 mm.
To examine the possibility of scaling up this reaction system, we carried out the esterification of L-leucine and n-butanol using two types of flow reactor, namely a desk-top-scale tubular reactor and a semi-bench-scale tubular reactor. These flow reactors exhibited sufficient productivity to confirm the potential of this system to be scaled-up. Our system also avoided the use of toxic solvents, long reaction times, and high temperatures, and so could be considered suitable for use in industrial applications. Although the yields of the esters synthesized by the flow methods were not particularly high, studies are underway aimed at improving the efficiency of the reaction and examining the application of this process to other amino acid esterification reactions.
Acknowledgements
The authors thank Dr Hidetoshi Miura of Chemiclea Inc. for discussions regarding the esterification of amino acids.