

1. Introduction
Polypeptides are linear polymers, built of twenty different amino acids (or residues), with a defined residue sequence [1]. A residue consists of the intrinsic side-chain, attached to the central alpha-carbon, and the common backbone corresponding to the linearly connected nitrogen (with hydrogen), alpha-carbon (with hydrogen), and carbon (with oxygen). The side-chain of the simplest residue, glycine, has only one hydrogen. On the other hand, the side-chain of the most complex residue, tryptophan, includes two rings, consisting of one nitrogen, nine carbons, and eight hydrogen atoms. Because different residues have a common backbone structure, their side- chains determine their nature―hydrophilic or hydrophobic.
Protein is a polypeptide in a living organism [1]. For example, there are about one hundred thousand kinds of proteins in human body. The linear residue sequence of a protein is called the primary structure. The popular local conformations of backbones, such as alpha-helix and beta-sheet, of proteins correspond to the secondary structure. Each kind of protein has its unique three-dimensional structure, called the tertiary native structure. Information on the tertiary native structure of a protein is quite crucial in understanding its biological function and role. Understanding the processes between the primary structure and the tertiary native structure is one of the most important problems in modern science. Understanding these processes is particularly important in this post-genomic era.
One of the most regular conformations adopted by proteins is the beta-sheet. Its basic unit is the (fully extended) beta-strand which is not stable itself. Because of this unstability of a single beta-strand, frequently leading to formation of amyloid fibrils and various fatal diseases, it is difficult to understand the formation processes and stability of proteins with beta-strands. Betanova [2] is a monomeric, beta-sheet protein consisting of three antiparallel beta-strands (Figure 1). Betanova has twenty residues, and its primary structure is given by Arg-Gly-Trp-Ser-Val-Gln-Asn-Gly-Lys-Tyr-Thr-Asn-Asn-Gly-Lys-Thr-Thr-Glu-Gly-Arg. In this work, we study the pathways between the folded native structure and unfolded conformations of betanova using UNRES force field [3] and the most popular computer simulation method, Metropolis Monte Carlo algorithm [4].
2. UNRES Force Field
In the united-residue (UNRES) force field [3] where a protein chain is represented by a sequence of alpha-carbon (Cα) atoms linked by virtual bonds with attached united side-chains (SC) and united peptide groups (p) located in the middle between the consecutive Cα’s (Figure 2). All the virtual bond lengths are fixed: the Cα - Cα distance is taken as 3.8 Å, and Cα-SC distances are given for each amino acid type. The energy of a protein in the UNRES force field is given by
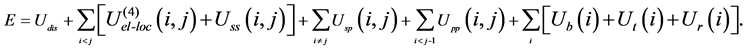 (1)
(1)
![]()
Figure 1. Tertiary native structure of betanova with three-stranded beta- sheet.
![]()
Figure 2. United-residue (UNRES) representation of a polypeptide. The interaction sites are side chain ellipsoids of different sizes (SC) and peptide-bond centers (p) indicated by shaded circles, whereas the alpha-carbon atoms (small open circles) are introduced to define the backbone-local interaction sites and to assist in defining the geometry.
Here, Udis denotes the energy term which forces two cysteine residues to form a disulfide bridge. The four- body interaction term U(4)el-loc results from the cumulant expansion of the restricted free energy of the protein. Uss(i,j) represents the mean free energy of the hydrophobic (hydrophilic) interaction between the side-chains of residues i and j, which is expressed by Lennard-Jones potential, Usp(i,j) corresponds to the excluded-volume interaction between the side-chain of residue i and the peptide group of residue j, and the potential Upp(i,j) accounts for the electrostatic interaction between the peptide groups of residues i and j. The terms Ub(i), Ut(i) and Ur(i) denote the short-range interactions, corresponding to the energies of virtual angle bending, virtual dihedral angle torsions, and side-chain rotamers, respectively.
The parameters of the UNRES force field were simultaneously optimized for four proteins of betanova (20 residues, three-stranded beta-sheet), zink-finger based 1fsd (28 residues, one beta-hairpin and one alpha-helix), HP-36 (36 residues, three alpha-helix bundle), and fragment B of staphylococcal protein A (46 residues, three alpha-helix bundle). The low-lying local-energy minima for these proteins were found by the conformational space annealing method. The parameters were modified in such a way that the native-like conformations are energetically more favored than the others. The global minimum-energy conformations found using the optimized force field are of the root-mean-square deviation (RMSD) values 1.5 Å, 1.7 Å, 1.7 Å and 1.9 Å from the experimental structures for betanova, 1fsd, HP-36 and protein A, respectively. After the parameter optimization, one set of the parameters is obtained for four proteins. The optimized parameters are not overfitted to the four proteins, but are transferable to other proteins to some extent. In [3], the procedures to obtain the optimized parameters used in this work are described in detail.
3. Monte Carlo Simulation
In the UNRES force field there are two backbone angles and two side-chain angles (see Figure 2) per residue (no side-chains for glycines). The values of these angles are perturbed one at a time, typically about 15˚, and the backbone angles are chosen three times more frequently than the side-chain angles. The perturbed conformation is accepted according to the change in the energy, following Metropolis Monte Carlo algorithm [4]. Since only small angle changes are allowed one at a time, the resulting Monte Carlo dynamics can be viewed as equivalent to the real dynamics [5]. For betanova, 100 independent computer simulations with 105 Monte Carlo steps (shortly, MCS) for each run were performed at a fixed temperature. We divided 105 MCS into 28 intervals (first ten 102 MCS, subsequent nine 103 MCS, and the next nine 104 MCS), and took average over conformations in each interval. These averages were again averaged over 100 independent simulations starting from the folded native structure.
During all simulations the values of RMSD from the experimental structure and the radius of gyration Rg were calculated using Cα coordinates. The fraction of the native contacts ρ was also measured during all simulations [5]. The values of ρ = 1 and ρ = 0 mean the folded native structure and a completely disordered conformation, respectively. RMSD, the radius of gyration, and the fraction of native contacts are the most important reaction coordinates in understanding the processes between the primary structure and the tertiary native structure.
Figure 3 shows the pathways between the folded native structure and unfolded conformations in the reaction coordinates ρ, Rg (in units of Å), and RMSD (in units of Å) for betanova at two different temperatures T = 100 (arbitrary units) and T = 200. As shown in the figure, the pathways are roughly linear. It is noted that they are similar even for quite different temperatures. In particular, up to the point of ρ = 0.7, Rg = 8.5 Å, and RMSD = 3 Å, the pathways are almost identical. The value of ρ = 0.7, Rg = 8.5 Å, and RMSD = 3 Å corresponds to a native-like conformation of betanova, still maintaining the antiparallel three-stranded beta-sheet. Therefore, the beta-sheet conformation of betanova is somewhat stable even in high temperature. And the pathways near the folded native structure of betanova less depends on temperature.
For T = 100, the pathway of betanova ends around the point of ρ = 0.2, Rg = 11 Å, and RMSD = 8 Å, corresponding to a disordered coil conformation. For T = 200, the pathway moves up to the point of ρ = 0.05, Rg = 12 Å, and RMSD = 9 Å. As expected, in high temperature, betanova is more disordered and extended.
4. Conclusion
Understanding the pathways between the folded native structure and unfolded conformations of a protein is one of the most important problems in this post-genomic era. Betanova is a monomeric, three-stranded antiparallel beta-sheet protein with twenty residues. The pathways between the folded native structure and unfolded confor-
![]()
![]()
![]()
Figure 3. Pathways between the folded native structure and unfolded conformations in the reaction coordinates (the fraction of native contacts ρ, the radius of gyration, and the root-mean-square deviation) for betanova. The pathways are obtained from 100 independent computer simulations at a fixed temperature. Black circles and continuous line show T = 100 simulation results, while red squares and dashed line represent T = 200 results. The values of ρ = 1 and ρ = 0 mean the folded native structure and a completely disordered conformation, respectively.
mations of betanova are studied using UNRES force field and the most popular computer simulation method, Metropolis Monte Carlo algorithm. At a fixed temperature, 100 independent Monte Carlo simulations are performed, starting from the folded native structure, and the pathways are generated at two different temperatures. The common and different characteristics for two betanova pathways are obtained in the reaction coordinates such as the fraction of native contacts, the radius of gyration, and the root-mean-square deviation.
Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (grant number NRF-2014R1A1A2056127).