The Unexpected Existence of Coding and Non-Coding Fragments along the Eukaryotic Gene ()
1. Introduction
Initial contributions established that, while the Sea urchin DNA contains the sole 5-methylcytosine (m5C) [1] , the E. coli DNA contains N6-methyladenine (m6A) and m5C [2] [3] .
The finding of these two classes of post-synthetically modified nitrogen bases along the bacterial heteroduplex was accompanied by basic observations dealing with the restriction-modification enzyme system [4] -[6] : m6A was shown to be due in part to the activity of the deoxyadenylate (dam) DNA methyltransferases (DNAmets) [7] -[9] and in part to the activity of type I, type II and type III methylating restriction-modification endonucleases involved in the digestion of the infecting m6A-free phage DNA [10] -[13] ; m5C was found to be due in part to the activity of the deoxycytidylate (dcm) DNAmets [9] [14] [15] and in part to the methylase activity of the sole type II restriction-modification system involved in the digestion of the infecting m5C-free phage DNA [11] .
The presence of m5C along the animal double helix was known earlier [16] [17] , and a small amount of m6A was soon found in DNA of protozoa [18] [19] , fungi [20] , algae [21] and higher vegetables [22] . The analysis of base composition in DNA of Drosophila was contradictory: one technique suggested an absolute absence of m5C [23] ; another showed that, in some of its sequences, C was still modified [24] [25] . Later a modest presence of m5C in the genome of insects was correlated with lethality [26] -[28] . In any case, it was established that, if in animal genomes, 1 to 5 out of every 100 Cs are modified [29] [30] , in plant genomes, this amount could increase up to 10 or even 30 per cent [31] [32] .
Our laboratory at the International Institute of Genetics and Biophysics (IIGB) of the National Research Council (CNR), in Naples, got interested in the topic because of studies on structuristics of rare nucleotides found in ox brain [33] [34] , plus and minus strand poliovirus RNAs synthesized at hourly intervals through the HeLa mitotic cycle [35] [36] and the nature of a high-molecular weight RNA extracted with hot phenol from nerve cells [37] [38] pertaining to the origin of mRNA. On the other hand, at variance with the polygenic structure of the operon regulating protein biosynthesis in bacteria [39] , while tentatively an operon organization was thought to act in higher organisms too [40] , the results collected by us at the Albert Einstein College of Medicine, in New York, on the amphi-directional regulation of a reversible reaction common to two inverted enzyme sequences [41] [42] accounted for a monogenic function of the transcriptional unit acting in mammals.
These points―apparently distant from each other―converged into an intriguing macromolecular manipulation, shedding light on the internal structure of the eukaryotic gene [46] . At the beginning of the 70s, in fact, while attention was paid to methylation of specific DNA sequences [47] , a problematic discussion focused not only on the origin and size of the eukaryotic pre-mRNAs and mRNAs [37] [73] [74] but consequently on the design of the transcriptional unit in higher cells [75] [76] . The background experiment, performed to elucidate both these aspects, originated from previous knowledge regarding the timing of DNA methylation during the cellular cycle [47] [67] and from the non-random genetic scattering of m5C along a semiconservatively replicating DNA chain [46] . This experiment showed a preferential methylation of gene promoter [46] (as reviewed in [88] ) and of all regulatory and signal sequences that do not code for mRNAs [46] (as reviewed in [89] ). In brief, in HeLa cells, if half of the population of hybrids between genomic DNA fragments of about 1 × 106 Daltons (used as “probes”) and pre-processed high molecular weight mRNAs (purified from nuclei) contained a large number of m5Cs, the whole population of hybrids between genomic DNA fragments of about 1 × 106 Daltons (also used as “probes”) and processed low molecular weight mRNAs (purified from polysomes) contained few, if any, m5Cs [46] . Since polysomal mRNAs were known to be much shorter than nuclear pre-mRNAs [75] [76] [111] , these results suggested for the first time that, at variance with the structure of bacterial genes (in an operon the cistrons were known to be constituted of coding sequences [39] ), the eukaryotic gene had to be thought of as a repetition, after the promoter, of intermittent “coding” and “uncoding” regions [46] [93] . For the sake of clarity, the DNA tracts containing methylated bases essentially were left out by mRNA [46] , namely methylation did not significantly involve the coding regions: methylation preferentially involved the uncoding ones, complementary to parts of pre-mRNAs to be removed during processing [46] . Three years later, Mandel and Chambon supported this finding [116] . Their electron microscopic pictures showed how a cDNA intermittently excludes segments of the corresponding pre-mRNA from hybridization: the sequences of pre-mRNA, non-hy- bridized with cDNA [116] , were nothing but the uncoding sequences [46] (Crick [87] and Gilbert [86] proposed to name them “introns”); the sequences of pre-mRNA, hybridized with cDNA [116] , were nothing but the coding sequences [46] (Crick [87] and Gilbert [86] suggested to call them “exons”).
The objective of this essay is to celebrate the eukaryotic gene fragmentation discovery and to emphasize how complex was the early 70s basic biochemical research promoting the modern Epigenetics and Genetic Engineering (as recognized in [150] -[160] ). Our next investigation will concentrate on the physiological evolution of coding vs. non-coding pre-messenger RNA regions. The exon and intron methylation differences will be evaluated as a function of the vertebrate evolution, while the perturbed regulation of transcription will be investigated in mammals in relation to the radiation-induced neoplastic consequences of the “repair-modification” activity.
2. The Pathway Using Methionine to Synthesize DNA Remained Unchanged in Phylogeny, While the Pathway Using Methionine to Modify DNA Evolved from Bacteria to Humans
Let us begin with description of the biochemical reactions lying on the basis of DNA polymerization and modification. The study was initiated in HeLa cells [46] [47] and completed in E. coli [48] [49] . However, coherently with evolution, it seemed appropriate to first treat the results regarding prokaryotes and, then, those regarding eukaryotes.
2.1. Synthesis and Methylation of DNA in Prokaryotes
One of the main purposes of this part of analysis was to verify if E. coli would be able to employ Met as a sole common donor of atoms for DNA synthesis and methylation, as HeLa cells do [47] [50] ; another purpose was to verify whether, on bacterial DNA, the timing for optimal yield of m5C would coincide with that for optimal yield of m6A [48] [49] .
It emerged that, when percolated through reverse phase HPLC [51] , control nitrogen bases, mixed in equimolar proportions, were eluted in the following time-order: C, m5C, G, T, A and m6A (Figure 1(a)). But there was a striking quantitative difference between the eluted unmethylated vs. methylated bases, when these were constituents of an E. coli DNA hydrolyzate: C, G, T, and A resulted in equimolar proportions, as expected; m5C and m6A were present in very small amounts (Figure 1(b)). Figure 1(c) illustrated the profile of an E. coli DNA hydrolyzate labelled with [14C]methyl-L-methionine ([14C]Met) in mid-culture growth cycle (CGC): C was the only base not incorporating the labelled carbon coming from the precursor; by contrast, m5C was labelled to a notable extent comparable to that of m6A. The radioactivity found in the two modified bases meant that post- synthetic DNA modification took place, as for HeLa cells [47] [50] , by virtue of transfer, via S-adenosyl-L-me- thionine (AdoMet), of the whole radioactive ?CH3 from [14C]Met to the DNA Cs and As. In addition, in Figure 1(c), the radioactivity found in the A, G and T bases revealed that, once again, as for humans [47] [50] , DNA synthesis emerged because there was, via C1-chain oxidation, an insertion of the labelled carbon coming from the ?CH3 of [14C]Met into the purine heterocycle of A and G and into the ?CH3 of T.
The quantitative analysis of the experiments performed through the CGC of E. coli (in the same conditions corresponding to those of Figure 1(c)), indicated that, while the curve of bacterial proliferation was S-shaped [48] , the synthetic and methylase pathways of DNA developed in a differential fashion (Figure 2(a)).
i. The specific labelling accounting for synthesis of A, G and T appeared to be much lower than that accounting for construction of m5C and m6A (this meant that the methylase pathway evolved faster than the synthetic pathway).
ii. The specific labelling of T was always lower than that of G and A since, one thought, the exit of the C1- chain, oriented towards synthesis of G and A, was highly facilitated in comparison with that oriented towards synthesis of T.
iii. The specific labelling of m5C was much lower than that of m6A, probably because the dam enzyme system worked faster than the dcm enzyme system.
![]()
Figure 1. Chromatographic analysis of a bacterial DNA hydrolyzate. Cultures of E. coli MRE 600 were grown on Petri dishes by inoculating 0.5 - 1 ml of minimal medium (containing 1 × 1011 cells/ml) into 100 ml of Luria-Bertani medium. At intervals of time during their CGC (roughly lasting 10 hrs), DNA was labelled for 40 min at 37˚C with 2 microCi/ml [14C]Met [48] . The 14C-labelled DNA was extracted with minor modification of the method described by Marmur [145] . A 30-micro- grams aliquot of purified DNA was introduced into a glass vial containing 300 microliters of 88% formic acid (75% final concentration). The vial was closed with flame and left at 160˚C for 1 hr [50] . The hydrolyzed residues were dried, suspended in 30 microliters of 20 mM trichloroacetic acid at pH 2.2 and run at 13,000 rev./min for 5 - 10 min in an Eppendorf centrifuge. A 20-microliters aliquot was percolated through a 131051 L ODS-55 Biorad Bio-Sil HPLC column (250 × 4 mm) in reverse phase condition [51] . Fractions of 0.4 ml were collected every 30 sec, while their content was resuspended in 50 microliters of water and tested for radioactivity in a Packard Tri-Carb TR beta-counter [48] [49] . (a) Free bases from a standard solution; (b) Bases present in 30 micrograms of an unlabelled DNA hydrolyzate extracted from a 2-ml suspension of 1 × 108 - 1 × 1010 bacteria maintained in minimal medium; (c) Bases present in 30 micrograms of a labelled DNA hydrolyzate obtained from a 2-ml suspension of 1 × 108 - 1 × 1010 bacteria maintained in Luria-Bertani medium. The left ordinate showed the concentration of bases detected in arbitrary units by a computerized instrument (continuous line); the right ordinate showed their radioactivity (dotted line). The analysis was repeated a dozen of times.
iv. In harmony with the development of the specific labelling of all nitrogen bases taken together and of the specific labelling of single A, G and T, the specific labelling of m6A gradually decreased during the CGC, when that of m5C rapidly decreased after the inoculum to rise again, with a sharp peak, in mid-CGC (an interpretation of these patterns was that the dam and dcm DNAmet activities are uncoupled) [48] [49] .
![]() (a)
(a)![]() (b)
(b)
Figure 2. Pathways leading to synthesis and post-synthetic modification of DNA. In E. coli [48] [49] , as in HeLa cells [47] [50] , to accomplish replication, the DNApol system exploited the dATP, dGTP and dTTP resulting from the C1-chain oxidation which used, via formic acid, the carbon coming from the ?CH3 of Met to build the purine heterocycle of A and G and the ?CH3 of T (this carbon did not enter the pyrimidine ring of C). (a) In bacteria, two parallel pathways led via AdoMet to post-synthetic DNA modification [48] [49] : on the one hand, the dam DNAmet system (including the methylation exerted by the type I, II and III RM endonucleases) constructed m6A on new DNA circles; on the other, the dcm DNAmet system (including the methylation exerted by type II restriction-modification endonucleases) constructed m5C on the same new DNA circles. (b) In Humans, Met also served as a common donor of atoms [46] [47] [138] : in the cytosol, the carbon coming from its ?CH3 through the C1-chain entered the purine ring of A and G and the ?CH3 of T; after formation of the corresponding dATP, dGTP, and dTTP and, after release of a PPi from each, in the nucleus the DNApol alfa [112] introduced, during S, the dAMP, dGMP, and dTMP into semiconservatively newly synthesized DNA chains, while Met catapulted via AdoMet its entire ?CH3 on given C residues located along these chains.
2.2. Synthesis and Methylation of DNA from the World of Bacteria to the World of Humans
The data reported above demonstrated that the two metabolic pathways, leading to synthesis and methylation of DNA, originated in the prokaryotic world by exploiting Met as donor of carbon atoms and ?CH3 groups (Figure 2(a)). This crucial property was preserved fairly well by the mammalian world (Figure 3(a) and Figure 3(b)), even if at the level of the DNA modifying enzyme bifurcation one of the two branches―the one yielding m6A on DNA (Figure 2(a))―was erased in HeLa cell DNA (Figure 2(b)).
![]()
Figure 3. Semiconservative transmission of m5C in human DNA. (a) Through an entire HeLa cell cycle [67] , the genomic DNA (labelled with [14C]Met) was hydrolyzed to bases and chromatographed: radioactivity in A, G and T (right ordinate) showed synthesis, while radioactivity in m5C (left ordinate) showed methylation [46] [47] . (b) During an entire mitotic cycle, nuclei isolated from HeLa cells were labelled with [3H-methyl]AdoMet and their DNA was hydrolyzed to bases and chromatographed: radioactivity in A, G, and T, accounting for synthesis, was negligible, while radioactivity in m5C showed methylation [47] ; (c) During S, HeLa cells were labelled with [14C]Met; then, after 14 hrs of growth in fresh medium (in the absence of radioisotope), their genomic DNA was hydrolyzed to bases and chromatographed: the measurement of radioactivity in m5C was repeated in the course of twelve CGCs. The insert suggested how, in the replication fork, methylation semiconservatively followed synthesis, since the labelled m5C per cell always decreased by half [109] . (d) Ultracentrifugation of genomic HeLa cell DNA in alkaline CsCl gradient [50] : the dashed line showed the m5C radioactivity (originating in [14C]Met) detected along the semiconservatively newly replicating chains, made heavier by a previous incorporation of BrdUrd; the solid line showed the OD at 256 nm of the separated lighter parental chains.
The abandonment of m6A by DNA of the majority of the highly evoluted cells was a consequence of the substitution, in them, of the obsolete bacterial antiphage restriction-modification defense [6] with the innovative immune reaction. Actually, with the extinction of the system of dam and dcm DNAmets [9] and of the system of restriction-modification endonucleases [11] -[13] , both characterizing the world of bacteria [52] , molecular selection led in mammals to the loss of m6A not only in nuclear [46] but also in mitochondrial [53] [54] and chloroplastic [55] DNAs. In these different nucleic acids, the m5C base was conserved by a novel DNAmet family that likely evolved through an alternate splicing [56] -[58] , as in mammals the fundamental defense against antigens was exerted by antibodies.
In such a structure, the question concerning the speed of reactions in the pathway leading to DNA synthesis vs. the speed of reactions in the pathway leading to post-synthetic DNA modification assumed great interest since, during the bacterial CGC, the rate of formation of T was always lower than that of A and G. Namely, the time required by the carbon, on the way from the ?CH3 of Met to the ?CH3 of T, was longer when compared to the time required for its insertion into the purine heterocycle of A and G [48] [49] . This differential rate was in harmony with the situation seen in HeLa cells: quantitatively, the biosynthetic rate of T was of the same order of that of A, even though a notable delay of its optimum with respect to the optimum of synthesis of A and G was observed (Figure 3(a)).
This kind of correspondence was expected, since either in bacteria (Figure 1) or mammals (Figure 3(a)) the process leading to synthesis of T had to join into a pyrimidine ring the carbamylphosphate and aspartate molecules before accepting, at position 5 of the newly formed U, the ?CH3 constructed with the carbon coming from formic acid. This carbon entered directly the A and G purine heterocycle (Figure 2(a)).
Biosynthesis of A, G and T transpired in comparable loci (in liquid matrix of a bacterial cell or in cytosol of a mammalian cell), whereas post-synthetic modification of DNA occurred in different compartments: in E. coli, it yielded m5C and m6A in the same liquid matrix (Figure 1(b) and Figure 1(c)); in HeLa cells, it yielded the sole m5C in the nucleus (Figure 3(b)). As follows, the longer time required by the nucleoside AdoMet to enter the nucleus would justify the advantage of the bacterial cell over the mammalian cell in exploiting the ?CH3 of Met: if in E. coli the specific building of A, G and T was much lower than that of m5C and m6A [48] [49] , in HeLa cells the specific building of A, G and T turned out to be either similar or only a little lower than that of m5C [47] [50] .
Summarizing, the large percentage of DNA methylation, necessary for antiphage defense in bacteria, became unnecessary in mammals.
Was this a non-turning point in evolution?
To make a long reasoning short: the prokaryotic world used Met to promote both the pathways leading to synthesis and methylation of DNA (Figure 1(b) Figure 1(c) and Figure 2(a)); this capacity was inherited almost completely by the mammalian world, with the exception that, in HeLa cells, the reactions yielding m6A on DNA were suppressed (Figure 3(a) and Figure 3(b)); the diversification of the speed of single reactions (in the pathways leading to synthesis and methylation of DNA) depended on the evolution of cellular organization, of course, since the eukaryotic nucleus concentrated in itself important genetic functions that were diffused earlier in the bacterial liquid matrix [47] -[49] .
3. In Humans the Cell-Cycle Methodology Served to Reveal Two Basic Laws Regulating DNA Methylation
Notwithstanding the exhaustive investigation on the evolution of the pathways leading to DNA synthesis and methylation, there were doubts in elucidating the biocatalytic processes dealing with the complex phenomenon of DNA methylation in eukaryotes: the existence of DNA demethylases, deaminases and reaminases was debated [59] -[61] ; the heterogeneity of the DNAmet system was revealed much later [56] [57] [62] -[64] . For this reason, in the early 70s, we preferred to investigate the timing of DNA methylation and consequently the precise macromolecular acceptors of the ?CH3 groups, namely the old or the new DNA chains: the experiments were made feasible by a method set up to determine the lengths of an entire mitotic cycle and its single G1, S, G2 and M phases [65] -[67] .
3.1. DNA Methylation Followed DNA Synthesis
It was suggested that DNA methylation would continue for some hours after DNA synthesis is completed [68] [69] . In this research, by incubating synchronized HeLa cells with [14C]Met used as common tracer for both DNA synthesis and methylation [47] , it was possible to verify that DNA methylation accompanied DNA synthesis: the labelled carbon atom, coming from the ?CH3 of Met, did not enter the pyrimidine ring but pierced via C1-chain the purine ring of A and G and the ?CH3 of T, while the entire ?CH3 of [14C]Met was transferred via AdoMet to DNA C (Figure 2(b)). Truly, the maximal labelling emerging from the 14C constituting the ?CH3 of [14C]Met was always found in S in four hydrolyzed bases: in A, G and T, implying synthesis, and in m5C, signifying methylation (Figure 3(a)).
3.2. Newly Replicating DNA Chains Were Semiconservatively Methylated
It was known that in isolated nuclei, in the absence of triphosphonucleosides (supplied by cytosol), DNA could not be synthesized. On this basis, by employing nuclei labelled with [14C-methyl]AdoMet, one could verify whether or not the absence of DNA replication would influence DNA methylation. The result was unexpected: in nuclei isolated from HeLa cells during S, among other DNA bases, only C continued to be methylated in the absence of DNA synthesis (Figure 3(b)). This showed that the two pathways of DNA replication and DNA methylation were separated from one another (Figure 2(b)): in whole cells, DNA methylation followed DNA replication during S (Figure 3(a)); in isolated nuclei, DNA methylation proceeded during S in the absence of DNA synthesis (Figure 3(b)). By itself the occurrence of DNA methylation in S-phase nuclei was not an absolute demonstration that it involved new chains formed just before their isolation; but the correlations between in vivo DNA synthesis and in vivo and in vitro DNA methylation strongly suggested this possibility [47] :
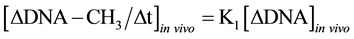
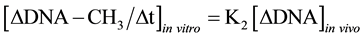
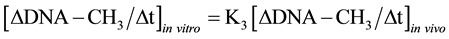
These equations established that the cells entering S would carry old chains inherited by parental cells, while targets of DNA met both in vivo and in vitro should be nothing but nascent chains: several of them still remained in the nuclei to be isolated [47] . A similar conclusion was reached thinking that, if old and new DNAs were methylated in isolated nuclei [70] , new DNAs would be formed without m5C in whole cells [71] .
Whatever the case would be, the semiconservativity of DNA methylation (Figure 3(c)), revealed by treating synchronized cells with the [14C]Met tracer [47] , was confirmed not only by the analysis that used restriction endonucleases [72] but also by the direct separation in alkaline CsCl of heavy bromodeoxyuridine (BrdUrd)- containing new chains and light non-containing BrdUrd old chains (Figure 3(d)).
4. Hybridizations of Pre-Processed Messenger RNA or Processed Messenger RNA with DNA “Probes” (Constructed during the S Phase by Shearing Newly Methylated DNA Filaments) Revealed an Alternation of Coding and Non-Coding Regions in the Human Gene
The hypotheses regarding the organization of the eukaryotic transcriptional unit had some analogy with those concerning the role of DNA methylation in higher organisms. From this, one deduced that the easiest way to directly characterize the eukaryotic gene design would be to exploit pure biochemical tools.
4.1. From Finding of Pre-mRNA and mRNA to Construction of DNA Probes
It is worth re-visiting in some more details the basic dispute into which we get involved. Georgyev and Mantieva found the polyribonucleotide called giant nuclear RNA in mouse [73] , as also evidenced by us in nerve cells [37] [38] . Penman, Vesco and Penman claimed that their polydisperse RNA had nothing to do with the giant nuclear RNA [74] . In return, Georgyev assumed that the polydisperse RNA would likely correspond to a product of maturation of the giant nuclear RNA [75] .
Accepting this assumption, Darnell, Jelinek and Molloy considered the giant nuclear RNA as pre-processed mRNA (measuring the size of a gene) and the polydisperse RNA as processed mRNA (much shorter than a gene): an agreement stating that a transcriptase would copy the whole eukaryotic gene to form a large pre- processed pre-mRNA, namely the heterogeneous nuclear RNA [76] .
This conclusion helped indeed to elucidate the structure of the monogenic transcriptional unit in mammals, since we judged such a monogenic unit as an extraordinary innovation exerted by evolution with respect to the complex bacterial polygenic transcriptional unit known as operon.
Therefore, the information reported above on semiconservative methylation of newly replicating DNA chains (Figure 3(c) and Figure 3(d)) served as a key for a crucial biochemical analysis [46] : segments of these chains, obtained through shearing, were employed as probes, labelled in m5C, in order to look for particular sequences located inside the eukaryotic gene (Figure 4(A)).
4.2. Our So-called “Pioneering Experiment on Gene Fragmentation” Demonstrated the Existence in the Human Transcriptional Unit of Coding vs. Non-Coding Regions
Filaments of semiconservatively newly made methylated DNA, previously separated during the S-phase in alkaline CsCl gradients (Figure 3(d)), were shortened through sonication into fragments of about 1 × 106 Daltons (Figure 4(A), left top) and hybridized with pre-mRNA (Figure 4(Ab)) extracted from HeLa cell nuclei [46] [66] [67] or with mRNA (Figure 4(Ac)) extracted from polysomes of these same cells [46] [77] : in the first case, the modified Cs were found in about 50 per cent of the hybrids “pre-mRNA/DNA probe” (signed with “plus” vs. “minus” in Figure 4(A)); in the second, the modified Cs were essentially absent in 100 percent of the hybrids “mRNA/DNA probe” (marked with “minus” in Figure 4(A)).
![]() (A)
(A)![]() (B)
(B)
Figure 4. Gene design. (A) Employing HeLa cells, semiconservatively newly made DNA chains, separated through CsCl in mid S-phase (a), served to construct by shearing methylated probes of 1 × 106 Daltons; their association with larger polyA- pre-processed messenger RNA (from nuclei) yielded methylated (50%) and unmethylated (50%) DNA/RNA hybrids (b); the same probes, associated with shorter polyA-containing processed messenger RNA (from polysomes), essentially yielded 100% unmethylated DNA/RNA hybrids (c). Actually, after the promoter, an alternation of coding and uncoding regions appeared to take place, since a preferential methylation characterized the same promoter and the sequences that did not code for mRNAs [46] [138] [147] . (B) The monogenic transcriptional unit was thought to imply an adjacency of 1-, 2-, n-blocs, each containing an initial methylated part and a final unmethylated part [46] [93] . The promoter (p) would correspond to the methylated part of the first bloc, followed by the first unmethylated coding (exon) part. Each following bloc should contain a methylated non-coding (intron) part and an unmethylated coding (exon) part. The last bloc should carry, at the end of its coding part (exon), the polyA signal and the stop codon(s). The RNApol II would synthesize pre-mRNA along the whole gene, while the pre-mRNA tracts―complementary to the methylated non-coding DNA sequences (from 2- to n-blocs)? would remain out of processed mRNA. (CAP) 5’-Methylated mRNA end; (pA) 3’-post-transcriptional polyadenylated pre- mRNA end.
For clarity’s sake, the result of the former experiment signified that the m5Cs were located along the promoter and along the non-coding regions inside the gene (all uncoding sequences, including the promoter, being complementary to regions to be removed from the unprocessed pre-mRNA); by contrast, the result of the latter experiment suggested that the joined translatable sequences of processed mRNA were complementary to the unmethylated coding sequences scattered in the gene.
Such a design of human transcriptional unit-structured in alternating non-informative and informative sequences, starting from the promoter (Figure 4(A), right top)―was illustrated at the 9th Meeting of the Federation of the European Biochemical Societies held in Budapest in 1974: the oral presentation was preceded by an invited paper that opened the ritual Special Issue of FEBS-Letters [46] .
Three years later, in 1977, the novelty brought by this paper was confirmed in France by Breathnach, Mandel and Chambon [78] : in their electron microscopic observations, the ovalbumin mRNA repeatedly did not hybridize sequences of the ovalbumin gene, forming characteristic loops (these were nothing else but the uncoding regions found earlier by us); differently, the sequences of the ovalbumin gene, that did become hybridized to the ovalbumin mRNA, were nothing else but our coding regions.
Soon, a number of other laboratories, mostly in USA, observed that the eukaryotic gene looked split: the pre-mRNA coding sequences were interrupted by uncoding ones [79] -[82] ; uncoding sequences were seen even in the adenovirus mRNA [83] [84] .
Then, when an intervening sequence was also described in the mouse beta-globin gene [85] , Gilbert [86] and Crick [87] proposed to call exons and introns, respectively, the coding and non-coding regions found in Naples [46] .
Nevertheless, while two years earlier the proposal of these new appellations Maclean and Hilder [88] , from United Kingdom, in the International Review of Cytology, attributed to us the priority of discovery of the “preferential methylation of bases in the promoter proximal region of the transcriptional unit”, two years later the introduction of these new appellations, Tentravhi, Guntaka, Erlanger and Miller [89] , from United States, in the Proceedings of the National Academy of Sciences of USA, attributed to us also the priority of discovery of the “preferential methylation of the early replicating regulatory sequences that do not code for mRNAs”.
In sum, if in HeLa cells half of the population of hybrids between genomic DNA fragments (used as probes) and pre-processed mRNAs (purified from nuclei) contained a large number of m5Cs (Figure 4(Ab)), the whole population of hybrids between genomic DNA fragments (used as probes) and processed mRNAs (purified from polysomes) contained few, if any, m5Cs (Figure 4(Ac)). Since polysomal mRNAs were known to be much shorter than nuclear pre-mRNAs, these results suggested that, in comparison with the bacterial operon structure (containing cistrons essentially representing informative sequences), the eukaryotic gene had to be thought of as a repetition, after the promoter, of alternate coding and uncoding regions (Figure 4(A), top right): methylation did not significantly involve the coding regions but involved the non-coding ones, complementary to parts of pre-mRNAs to be removed during processing [46] .
5. Evolution Combined a Variable Number of Coding-Uncoding Modules in Eukaryotic Genes Depending on the Lengths of the Polypeptides to Be Expressed
Following the demonstration that the infection by a lytic virus, like poliovirus, was found to be rigorously cell- cycle dependent [35] [36] , the observation of the design of the eukaryotic transcriptional unit [46] induced to investigate the site of recombination between the genome of an oncovirus, like SV40, and that of a host-cell, like mouse fibroblasts (the oncogenome integration needing at least an uncoding-coding module as specific target).
The working hypothesis was based on the assumption of two possible mechanisms of virus-induced cell transformation: one involving a complete loss of cell regulation and the other involving a partial loss of cell regulation. In the first instance, the virus DNA would be integrated between the uncoding and coding regions of the module while, in the second, the virus DNA would be integrated in the uncoding region only [90] .
The genetic engineering experiments spoke in favor of the former mechanism, being facilitated during the S- phase [91] [92] . This mechanism―in addition to the information carried by the SV40 early gene―exploited the information carried by an adjacent short piece of host-cell DNA to yield the so-called non-truncated, i.e. entire (fused), T-antigen: the truncated T-antigen, encoded by the SV40 early gene, did not contain the signals for polyadenylation and stop codons; these signals were brought by the joined host-cell DNA piece [93] .
These new acquisitions led us to carefully consider the question, posed by Gilbert [86] , on why the genes are broken down into pieces.
The origin and evolution of the uncoding-coding structures seemed problematic to him, not only because both the “exon theory of genes” and the “insertional theory of introns” debated the presence or absence of uncoding sequences in “primordial genes”, but also because the statistical analysis assumed that an “exon shuffling” played an important role in the origin of either ancient or modern genes.
Despite the fact that we did agree with such a viewpoint, our attention was focused, rather, on the order of the two classes of regions in a module. Why, we questioned, an uncoding region should precede a coding one, and not vice versa?
Independently from the concept of exon shuffling, one would answer that transcription starts in any case from a promoter.
A modular gene organization should be in good harmony with this need: in the initial module, the promoter should play the role of an uncoding region, preceding the first exon; in the last module, after the intron, an exon should necessarily carry an end with signals for polyadenylation and stop codons (Figure 4(B)). Thus, inside the gene, the promoter and the various non-coding regions should exert a regulatory function, while the various coding regions should carry information for a given protein [46] .
But were the ?CH3 groups (preferentially carried by the promoter and non-coding regions along the gene) correlable indeed with the genetic regulation of transcription?
A circumstantial attention to this correlation will be paid below.
Allow us emphasize the fact that the association of non-coding regions with methylation vs. the practical absence of methylation in coding regions (Figure 4(B)) represented ideal properties useful to evaluate the “age” of the regions themselves, i.e. if unmethylated coding sequences should be considered phylogenetically older while methylated non-coding sequences should be considered phylogenetically younger.
From the viewpoint of evolution, we assumed that the unmethylated coding regions probably became ‘conservative’ because the conventional excision-repair system always reconstructed them by maintaining any possibility of error at a minimum (e. g. the high conservation of genes coding for histones), as the continuous repetition of possible incomplete repair of the methylated uncoding sequences―through a long chain of cell cycles [90] ―probably caused their wide variability.
On the basis of this variability selection would have operated by creating (through ad hoc transpositions) new transcriptional units containing non-coding regulatory and signal sequences [91] [93] -[96] .
An insight of this kind might also explain why the amount of the uncoding portion in many eukaryotic genes was found to be so large when compared with that of their coding part, being the proportion dealing with the lengths of the proteins to be made, of course [53] . After all, the chain of biocatalytical reactions able to join in sequence various uncoding-coding modules to form the genes expressing these proteins had not yet been understood.
6. The Alternation of Coding and Non-Coding Regions in Eukaryotic Genes Was Convalidated by Their Restriction Minimaps Showing a Scattered Distribution of the Methylatable Sites
At the 7th International Symposium on Macromolecules in the Functioning Cell, held in Taormina in 1990, my talk entitled “Is 5-methylcytosine a regulatory signal in eukaryotic gene expression?” emphasized the concept of split eukaryotic gene [46] analyzing its methylatable sites recognized by restriction-modification endonucleases [97] . Then, the split design of the gene, observed in HeLa cells, that I extensively elucidated in research seminars given at the King College of London, in 1991, and at the Cancer Center of the University of Texas, Houston, in 1992, contributed to motivate at the “Incubator of Ideas” of Stony Brook, USA, in 1993, the project of a long-term biotechnological co-operation between the Italian CNR and the State of New York [98] , while a review on “5-methylcytosine in genes with methylation-dependent regulation” opened an issue of FEBS-Letters [99] .
Complementing this, it is not superfluous to further make the subject clearer through a careful reading of restriction minimaps constructed for a large number of human genes regulated by m5C: their genetically discontinuous “geometry” (Figure 4(A)) exhibited, after the promoter, a sui generis wavy chain of hypomethylatable coding and hypermethylatable uncoding regions (Figure 4(B) and Figure 5). Both in housekeeping (HK) and tissuespecific (TS) transcriptional units, they were roughly contained beneath the limits of a “scalene triangle”.
For example, the calcitonin (CALC) transcriptional unit?representative of about thirty fully A, T, G, C-se- quenced and m5C-regulated TSs―was heavily methylatable upstream and slightly methylatable downstream (Figure 5(b)).
In such a framework, at least four pairs of dinucleotides behaved as targets recognizable by restriction-mod- ification endonucleases [98] [99] .
In exons, two of them were monomethylatable:
5’-Tpm5C-3’ 5’-Cpm5C-3’
3’-ApG-5’ 3’-GpG-5’
![]() (a)
(a)![]() (b)
(b)
Figure 5. Scattered methylatable cites along the HK and TS genes in mammals. The restriction minimaps were drawn using the Mapdrow DNAstar Program. At the 5’-end of a gene, the promoter region showed the highest number of dcm-sites, accounting for a significant hypermethylation [98] [99] [146] . Going from 5’ to 3’, while the alternate coding (exon) sequences carried a relatively low number of dcm-sites, the alternate uncoding (intron) sequences carried a relatively high number of dcm-sites. However, this level steadily decreased going to the 3’-end. (a) The HK ODC gene contained 9.043 nucleotide pairs; (b) The TS CALC gene contained 7.637 nucleotide pairs. (Ends) dcm-inhibited restriction endonucleases; (1) single dcm-sites; (2-6) multiple dcm-sites.
In promoter and introns, the other two were dimethylatable:
5’-m5CpG-3’ 5’-Gpm5C-3’
3’-Gpm5C-5’ 3’-m5CpG-5’
At any rate, the average quantitative distribution of these pairs?which corresponded to those isolated from the DNAse I digests [100] -[102] ―was similar in HKs (Figure 5(a)) and TSs (Figure 5(b)), despite the fact that in the case of TSs (going in the direction from 5’ to 3’) the monomethylatable dinucleotide pairs proportionally increased while the dimethylatable dinucleotide pairs proportionally decreased [98] [99] .
Thanks to identification of all these methylatable “words”, the promising 1993 seemed to anticipate, therefore, that the biomolecular studies were moving into a challenging new cycle, whilst new resources such as restriction gene minimaps were enabling genome-wide investigations that could potentially identify most common genetic determinants of human health, disease or drug design.
7. The “Repair-Modification” System Played a Role in Either Gene Evolution or Neoplastic Cell Transformation
If we look back for a moment at the investigations regarding the cell-cycle dependence of DNA methylation [47] vs. the non-random scattering of m5C along semiconservatively newly replicating DNA chains [46] , we can appreciate how they not only paved the way to the discovery of the hypermethylation of promoter and all regulatory and signal sequences that, in the eukaryotic gene, did not code for mRNAs [46] but also considered the corollary experiments which, albeit at the beginning oriented towards other goals (particularly towards deciphering of the ?CH3 language), at the end helped to shed some new light on the fine control of post-synthetic DNA modification.
7.1. A Differential Methylation of Euchromatic vs. Heterochromatic DNAs Took Place
Evidence indicated that the S-phase should be subdivided into two parts with respect to the characteristics of DNA, since early replicating euchromatic DNA tended to be CG-rich, while late replicating heterochromatic DNA tended to be AT-rich [103] .
Furthermore, the DNA fraction extracted from Chinese hamster cells in early S was methylated to a greater extent when compared to the DNA fraction extracted from the same cells in late S [104] .
This difference was reproduced in HeLa cells [46] . On its basis, we supposed that, while in newly replicating DNA chains there would be sequences not uniformly methylated in the course of S, the CG-rich sequences would be preferentially methylated [46] .
7.2. A Biological Clock Controlled Methylation of Specific Replicating Sequences
Since DNA did not appear to be uniformly methylated in S, investigations continued to focus on nitrogen bases specifically adjacent to m5C [46] rather than considering the rough subdivision of hypermethylated euchromatic vs. hypomethylated heterochromatic replicons [104] .
With this purpose in mind, methylated DNA coming from HeLa cells was fractioned in alkaline Ag+/Cs2SO4 gradients: a small heavy CG-rich fraction and a large light AT-rich fraction were obtained (the first―containing genes for rRNA―was mainly expressed in early S and, the second, was mainly expressed in late S); as for the m5C concentration, if in early S it was found to increase on the heavier peak, in late S it was found to increase on the lighter peak [105] -[107] .
All this suggested that, at the different stages of S, one dealt with CG- or AT-rich regions probably polymerized in correspondence of specific hypo- or hypermethylated templates: diversely, it seemed that the genes would be methylated according to a given order and intensity along the newly replicating chains [105] .
This idea was definitely demonstrated when we observed that the foldback DNAs were subjected to hypermethylation: if in HeLa cells―through the S-phase―a methylation wave chronologically involved palindromic, highly repeated and moderately repeated sequences, the unique sequences were characterized by a minimal late methylation [108] .
7.3. Both CG- and AT-Rich Sequences Were Targets for Methylation
A circumstantial analysis of the two genomic DNA fractions, separated in alkaline Ag+/Cs2SO4 gradients during S, confirmed that m5C had a differential distribution in them: the highest concentration of methylated sequences was found in the denser side of the heavy fraction; in the light DNA the highest concentration of methylated sequences was found in the lighter side [105] -[107] .
This implied two generalizations: hypermethylation on CG-rich sequences would offer no surprise because of its rather statistical character (the CG-rich rRNA genes would be hypermethylated according to the size of their CG targets); hypermethylation on AT-rich sequences would seem to work, instead, against the principle of CG target size, since a decrease in C residues would correspond to an increase in their methylation. Hence, when the extra-S time was characterized by a minimal, probably de novo DNA methylation, S exhibited maintenance DNA methylation [47] .
For this reason, one assumed, the mechanisms discussed above should function in S [90] .
But it was obvious that, with consecutive cell cycles (Figure 6(a)), the methylated sequences could not increase without limits [109] , also because changes in the amount of methylated DNA during development [100]
![]() (a) (b)
(a) (b)
Figure 6. Inverse correlation between DNA methylation and gene expression as a function of cell cycle or repair-modifica- tion. (a) DNA methylation [47] vs. biosynthesis of DNA [101] , RNA [110] , and protein [111] : the hours were signed by points where spokes crossed the circular “abscissa”; the ordinates were represented by the same spokes; the white arrow, against M, referred to a line of cell-cycle asymmetry L [65] [67] . Following appropriate labelling, at hourly intervals, macromolecules were extracted, purified and analyzed on gradients [50] [101] [110] [111] . Replicative synthesis of genomic DNA (__) and its methylation (--) showed apogees in S, as in Figures 3(a)-(b), while repair synthesis of genomic DNA (--) was rather constant around the cycle [50] [90] [138] . Replication of mtDNA (__) showed maxima in S and G2, whereas its methylation also occurred in S [54] [101] . Differently, transcription of RNA (__) and its translation into protein (_ _) exhibited almost overlapped apogees in G1 and G2 [90] [138] . In M, all macromolecular biosyntheses were rather repressed [35] [36] [90] [138] . (b) Repair-modification mechanism [53] [95] [138] [142] -[144] [148] : UV at random would induce the building of TT-dimers in vicinity of symmetrically dimethylated 5’-m5CpG-3’/3’-Gpm5C-5’ dinucleotide pairs, for instance (2); in this case, after digestion of damaged regions (3), which previously included a m5CpG dinucleotide (1), the cell cycle-inde- pendent excision-repair would replace the m5C nucleotide with a simple C nucleotide, since repair DNApol would not find any methylated dCTP in the soluble pool of triphosphonucleosides; the resultant hemi-methylated CpG/Gpm5C dinucleotide pairs, namely the incompletely reconstructed RPs (4), should gain the capability of resuming transcription; in S, the cell cycle-dependent DNAmet would add to C the lost ?CH3 (5), providing completely reconstructed RPs able to silence the transcription again.
and in differentiated tissues [29] [30] were not conspicuous.
So, whatever the mechanism of DNA methylation was in S, statistical or specific, it was reasonable to imagine that, mostly during the extra-S time, each small wave of de novo DNA methylation should be followed by a corresponding small wave of DNA de-methylating repair [53] .
7.4. After a Gene Radiodamage, the Complete Rebuilding of Unmethylated Coding Regions Was Guaranteed by “Excision-Repair”, While the Complete Rebuilding of Methylated Uncoding Regions Was Guaranteed by “Repair-Modification”
In HeLa cells, replication of genomic DNA [65] [67] [101] was followed by its methylation [47] in S, whereas transcription [110] and translation [111] mainly resulted in G1 and G2. This was the first concrete suggestion as to a macroscopic inverse correlation observable between gene methylation and expression (Figure 6(a)). As for the enzyme components: DNA polymerase (DNApol) alpha, responsible for replication of genomic DNA, was induced in S; DNApol gamma, responsible for replication of mtDNA, was made in S and G2; DNApol beta, involved in rebuilding, was produced at a rather low level through the whole mitotic cycle with some increase in early S (the increased DNApol beta repair activity in early S signifying an effort by the cell to highly reconstruct the genomic DNA just before its replication) [121] . Moreover, in conformity with the sharp peak of maintenance genomic DNA methylation in S (Figure 6(a)), the DNAmet system [63] was induced in S as well (Figure 3(b)), while a moderate presence of DNAmet characterized the rest of the cell cycle mainly accounting for de novo genomic DNA methylation (Figure 3(b)). During S, a coupling―definable as repair-modification―took place between the activities of DNApol beta (involved in excision-repair) and DNAmet (making re-methylation), whereas during the extra-S time an uncoupling occurred between these two enzyme systems [53] [64] .
Thus, using synchronized HeLa cells [65] [67] , we took a look at the differential reconstruction of the UV- damaged unmethylated coding and methylated uncoding gene sequences (Figure 6(b)): in a repair patch (RP) located along given promoters or introns (1), it could happen that a symmetrically dimethylated 5’-m5CG- 3’/3’-Gm5C-5’ dinucleotide pair would flank a UV-induced TT-dimer (2); after digestion of the damaged nucleotide sequence (3), the excision-repair (being the methylated dCTP absent in the soluble pool of triphosphonucleosides) would replace―in the “incompletely” reconstructed sequence―the previous m5C nucleotide with a normal unmethylated cytosine nucleotide (4). Even if the existence of DNA de-methylating proteins should not be excluded in general [113] , this “non-enzymatic de-methylation” certainly would occur through the whole cell cycle. Anyhow, during S―via AdoMet―DNAmet would add to the incompletely reconstructed (unmethylated) DNA sequence (4) the lost ?CH3 group, rebuilding the previous symmetrically di-methylated 5’-m5CG-3’/3’- Gm5C-5’ dinucleotide pair (5). This “complete” rebuilding of a previously methylated RP would require, in any situation, a “re-methylation” catalyzed by DNAmet [53] [95] [114] , as demonstrated by the fact that the labelled carbon atom coming from the ?CH3 of [14C]Met (Figure 3 and Figure 2(b)) was found in the T, G, A and m5C nitrogen bases carried by the re-methylated RPs (Figure 7(c)) distributed along the light parental strands (Figure 7(b)). Yet, to what kind of regions―along the eukaryotic transcriptional unit―did such RPs belong?
Sedimentation of the semiconservatively newly replicating chains, made heavier in alkaline CsCl by BrdUrd incorporation (Figure 7(a)), was confronted with that of the labelled RPs appearing in the light parental chains (Figure 7(b)). These light parental chains, after an extensive shearing by sonication [46] , were hybridized with polyA-containing pre-mRNA extracted from nuclei (Figure 7(c)) or with polyA-containing mRNA extracted
![]()
Figure 7. Differential repair-modification of introns and exons following a DNA radiodamage [96] . (a) HeLa cells were labelled, in mid S-phase, for 1.5 hrs with 80 Ci/mmol [14C]Met in the presence of 10 mM BrdUrd; (b) HeLa cells were UV- irradiated for 45 sec and labelled with 80.0 Ci/mmol [14C]Met in the presence of 10 mM BrdUrd plus 5 mM hydroxyurea. In both cases, the CsCl gradients, brought to pH 8.5, were run in the rotor SW55Ti for 60 hrs at 32,000 rev./min. The [14C]-repaired parental DNA (b), sheared by sonication to fragments of about 1 × 106 Daltons, was hybridized with the [3H]-labelled pre-processed polyA-containing mRNA fraction, purified from nuclei (c), or with the [3H]-labelled processed polyA-containing mRNA fraction, purified from polysomes (d). Both pre-processed and processed messenger RNA fractions were labelled in unsynchronized cells with [3H]uridine for 30 min in the presence of 0.04 micrograms/ml Actinomycin D and purified through oligo-dT cellulose. The hybrids were hydrolyzed to bases and chromatographed by HPLC. The reduced scale at the right-hand side (14C-label) concerned m5C.
from polysomes (Figure 7(d)). In the first combination, the hybrids “DNA/pre-mRNA” showed an appreciable amount of m5Cs; in the second, the hybrids “DNA/mRNA” did not appear to be methylated. These results signified that re-methylation involved the uncoding parts of the repaired genes and that the coding parts of these genes only underwent a rebuilding of their primary base sequence, without modified bases.
In summary, we considered one of the possible mechanisms―repair-modification―through which the presence of m5C in eukaryotic genes might have exerted a key role in the evolution of their unmethylated coding vs. methylated uncoding regions: after radioinduced damage, the RPs formed in exons (where the modified bases were practically absent) would be completely reconstructed by the sole excision-repair, whereas the RPs formed in promoters and introns (where the modified bases were normally present) would be incompletely reconstructed by excision-repair. Only later, in S, the unmethylated RPs would become completely reconstructed by re-me- thylation.
We assumed, therefore, that the repair-modification machinery exerts a continuous differential pressure on evolution of given gene pieces, when methylated to a different extent (Figure 7(c) and Figure 7(d)).
In particular, according to Darwinian concepts, repair-modification would lead to a very high variability of introns (if hypermethylated), whereas on the basis of this wide variability selection would favor their transposition from one gene to another. In them, the conservative nature of exons (if unmethylated) would be ensured by excision-repair.
Besides, it is worth stressing the possible influence that the incomplete repair of RPs (see the de-methylation shown in Figure 6(b4)) could exert on resumption of transcription by virtue of the cell cycle-dependent inverse correlation seen between DNA methylation and RNA synthesis (Figure 6(a)).
In this case, in harmony with the azacytidine effect [94] observed in HeLa cells (Figure 8), certain previously silent (Figure 6(b1)) and then re-activated (Figure 6(b4)) genes would lead even to neoplasy.
After all, the idea suggesting that the radio-induced DNA de-methylation could be considered as a feasible molecular basis for cancerogenesis [53] conformed with the known fact denouncing that at least 30 per cent of tumors are really induced in humans by radiations [148] [149] .
![]()
Figure 8. Inverse correlation in S phase between inhibition of DNA methylation by 5-azacytidine and polyA-containing pre- mRNA transcription. The experiments were performed in synchronized HeLa cells, as for Figure 3(a) and Figure 6(a): following the inhibition of genomic DNA methylation by 5-azacytidine [94] , some resumption of transcription was evidenced (dashed area). The left ordinate showed the rates of pre-mRNA synthesis in control (black squares) and 5-azacytidine-treated (white squares) cells labelled with [3H]uridine; the right ordinate showed the rates of genomic DNA methylation in control (open circles) and 5-azacytidine-treated (closed circles) cells labelled with [14C]Met.
8. The Arduous Biochemical Approach to Decipher the Language of Gene Methylation in Eukaryotes Was Crowned with Success by Its Direct Bisulfite Reading
The finding of an alternation, after the promoter, of coding and uncoding regions in the eukaryotic transcriptional unit and the consequent demonstration that these regions are differentially methylated, or re-methylated, inevitably stimulated curiosity to decipher the language marked by their m5Cs.
8.1. At Variance with the Systems of Regulation of the Operon Activity in Bacteria, an Inverse Correlation between Gene Methylation and Transcription Was Shown in Humans
In bacterial DNA the m6A and m5C modified bases were related not only to the restriction-modification anti-phage defense [6] : complementing the induction or repression operon functions [39] , they were further associated with an auxiliary mechanism capable of partially regulating transcription in the same phage-infected microorganisms [115] . This “embryonic” stage of control evolved in the major part of eukaryotes: in them the higher concentration of m5C, present in the promoter and introns, was suggested to be responsible for modulation of gene expression [46] , as supported by others [60] [116] [117] and reviewed [89] [118] . Such an involvement found its first clear demonstration in the fact that the maximal rate of DNA methylation [47] [119] followed the maximal rate of DNA replication [101] during S, while the maximal rates of transcription [110] and translation [77] [111] appeared during G1 and G2 (Figure 6(a)). Further results supporting the idea about the role of m5C in regulation of gene expression were soon detected [120] -[123] , afresh to those on the methylation-de- pendent regulation of integrated viral genes [92] -[109] [117] [124] [125] . Moreover, maintenance DNAmet [47] [72] was shown to recognize hemimethylated 5’-CpG-3’/3’-Gpm5C-5’ dinucleotide pairs to yield dimethylated 5’-m5CpG-3’/3’-Gpm5C-5’ dinucleotide pairs (Figure 3(c)), whilst an inverse correlation between methylation and expression was established for a large number of sequenced HK and TS genes [98] [99] . In fact, an inverse correlation between gene methylation and transcription did occur in animals signifying, at the same time, that it could be caused most likely by the higher concentration of m5C in promoter and intron sequences: one theory justifies the other, and vice versa.
8.2. A Study Focused on Isolation of Specific Targets for Methylation in Native Human DNA
After establishing that in eukaryotes the ?CH3 groups are preferentially attached to the CpG dinucleotides [106] [107] [126] , as in bacteria [127] , clearcut evidence about the existence of two classes of targets for DNAmet emerged, as mentioned already, from analysis of HeLa cell double-helical DNA in alkaline Ag+/Cs2SO4 [106] .
This type of gradient separated a heavier fraction (representing about 20% of the total) from a lighter one (representing about 80% of the total) [128] .
However, the consequent ultracentrifugation of these fractions in CsCl [106] further showed that the heavier, banding at 1.715 g/cm3, contained 53% CG (10% of the total CG), whereas the lighter, banding at 1.703 g/cm3, contained 40% CG (32% of the total CG).
Generalizing, four possible triplets as sites for DNAmet recognition were suggested: GCG and CCG, in CG-rich sequences; ACA and ACG, in AT-rich sequences [107] .
The evidence that the specific methylation of CG-rich sequences (as those for rRNAs) was maximal in early S and that the specific methylation of AT-rich sequences was maximal in late S not only demonstrated that the DNA sequences were replicated and methylated following an order during S [107] [108] but also pointed that the DNAmet activity exerted the role of maintenance modification along CG-rich sequences and the role of de novo modification along AT-rich sequences [107] .
This important deduction was in accordance with the fact that the extra-S methylation (about 10% of the total) involved some AT-rich sequences either along the old or the new chains, when the S phase methylation (about 90% of the total) almost exclusively involved newly replicating chains [47] [50] .
In S, the maintenance DNAmet function [63] followed at a distance of about 30 min [47] the DNApol alpha activity [112] .
The reasoning made sense because, in S, the maintenance modification―involving CpG targets in newly replicating chains―would be induced by the ?CH3 of the m5C present in the complementary strand as a genetically encoded signal.
Otherwise, a CpG dinucleotide (Figure 3(c)) would be methylated on the new chain in the presence of a complementary already methylated GpC dinucleotide on the old chain [47] [72] ; differently, during the extra-S time, a de novo modification occurring on an ApC dinucleotide, for instance, could not be induced by a signal coming from the complementary TpG dinucleotide since this cannot be methylated at all.
8.3. Both De Novo Monomethylated and Maintenance Dimethylated Dinucleotide Pairs Were Found in Humans
For the sake of clarity, the above-established sedimentation of CG- and AT-rich sequences in alkaline Ag+/ Cs2SO4 suggested the existence of at least four targets for methylation [106] :
GCG, CCG, ACA, and ACG.
In one of the trinucleotides there was a methylatable CpA; in three of them, the methylatable CpG was repeated. For this reason, a direct isolation of such restricted targets was required (Figure 9).
![]()
Figure 9. Methylated “words” in eukaryotic genomic DNA. Experiments with synchronized HeLa cells [65] [67] demonstrated that, in double helix (characterized by antiparallelism of complementary chains) two symmetrically dimethylated palindromic dinucleotide pairs and six asymmetrically monomethylated non-palindromic dinucleotide pairs were detected [102] [138] . DNA was labelled in isolated nuclei with [14C-methyl]AdoMet, as for Figure 3(b): after extraction, in the middle of each cell-cycle phase (G1, S, G2 and M), it was digested with pancreatic DNAse I [129] to chromatografically separate various dinucleotides [102] . The white columns show the dinucleotide molar concentration, expressed in OD at 256 nm; the black columns, expressed in DPM, show the radioactivity of their m5C.
This was achieved by exploiting DNAase I digests [126] [129] of DNA samples methylated in isolated nuclei of previously synchronized cells with [3H-methyl]AdoMet [47] [109] . Chromatographic analysis of the material contained in these digests [102] yielded a number of obviously unmethylated dinucleotides
ApA, ApG, ApT, CpT, and TpT
and, for the rest, four dinucleotides methylated to a different extent:
m5CpT, Cpm5C, m5CpA, and m5CpG.
Their methylation level was cell-cycle dependent, in agreement with [47] : in M, few CpGs were methylated; in G1 and G2, methylation of CpGs slightly increased, in comparison with that seen in M, and there appeared some methylation on CpTs, CpCs, and CpAs; in S, again, a methylation of four dinucleotides
m5CpTs, Cpm5Cs, m5CpAs, and m5CpGs
was detected. In this phase, methylation of CpGs became intensive (90% of the total), accounting for its maintenance character along newly made chains (Figure 3(c)). The occurrence of methylation on CpTs, CpCs, and CpAs accounted instead for a de novo phenomenon, which characterized especially the extra-S part of the interphase [47] . Considering the two possible directions of the separated dinucleotides, due to the double helix antiparallelism (the 5’-3’ and 3’-5’ directions could not be distinguished chromatographically), the analysis actually regarded the identification of eight words written in the genome in terms of dinucleotide pairs:
5’-m5CpG-3’ 5’-Tpm5C-3’ 5’-Cpm5C-3’ 5’-m5CpA-3’
5’-Gpm5C-3’ 3’-ApG-5’ 3’-GpG-5’ 3’-GpT-5’
+ + + +
3’-m5CpG-5’ 5’-m5CpT-3’ 5’-m5CpC-3’ 5’-Apm5C-3’
3’-Gpm5C-5’ 3’-GpA-5’ 3’-GpG-5’ 3’-TpG-5’
Briefly, if the CpG target only allowed a final dimethylation of dinucleotide pairs, as expected from semiconservative methylation [47] [72] [102] [130] , the other combinations always led to a monomethylation of dinucleotide pairs [102] . Still, what could be demonstrated about the diversity of the methylation code at the level of the promoter and at that of the intervening uncoding sequences?
8.4. In the Promoter of the Transglutaminase Gene the Bisulfite Sequencing of 5-Methylcytosine Showed Two Fully Methylated CpG-Rich Domains Close to the 5’-End and One Fully Unmethylated CpG-Rich Domain Close to the 3’-End
Using the model of human transglutaminase (hTGc) gene [131] and the method based on bisulfite conversion of C to T residues along a DNA filament [132] , the following analysis showed that m5C, behaving as bisulfite-in- dependent base [133] , could be sequenced directly despite its low molar proportion [134] .
The hTGc gene was chosen principally because it was among those that are regulated by methylation [135] . In its 1665 bp long promoter [131] , three CpG-rich domains were characterized (Figure 10): the first domain (330 bp), close to 5’, contained 12 CpGs corresponding to an average frequency of 3.63% with respect to the total number of nucleotides; the second (227 bp), roughly located in the middle of the promoter, contained eight CpGs corresponding to an average frequency of 3.52%; the third (264 bp), close to 3’ (including 70 bp of the 73 bp long 5’-UTR), contained 31 CpGs corresponding to an average frequency of 11.74% [136] .
In leukocytes and lymphocytes, where hTGc is silent [135] , out of the three CpG-rich domains, only the first two were found to be methylated, while the third, on the 3’-side, did not present any m5Cs: the 11 CpGs of domain 1 and the 7 CpGs of domain 2 were methylated 100%; the 31 CpGs of domain 3 were on the contrary unmethylated 100% (in this domain the 5’-UTR, also resulting 100% unmethylated, was almost entirely included) [136] .
The lack of m5C in domain 3 was expected, in agreement with the conventional definition of unmethylated CpG-rich “island” (where the intrafilament CpG/GpC ratio has to be higher than 0.6); but the occurrence of methylation in all CpGs of domains 1 and 2 was unexpected. Domain 1 contained 79 Cs and 11 CpGs out of 330 bases; domain 2 contained 44 Cs and 7 CpGs out of 227 bases; domain 3 contained 116 Cs and 31 CpGs out of 264 bases. Compared with the average 5%-methylation of human genomic DNA [137] , methylation of domains 1 and 2 corresponded to 15.18% and 18.18%, respectively [136] . This confirmed the idea that the promoter of an inactive gene should be characterized by hypermethylation [46] [98] [99] .
Certainly, HUVEC cells joined their hTGc gene activity with a loss of m5C along the hTGc promoter domain 1, at least at the −1380, −1349, −1338, and −1320 CpG sites [138] . This is an appropriate example of direct
Methylated Methylated Unmethylated
![]()
Figure 10. Scattered methylation along the repressed hTGc gene promoter in lymphocytes. The sequenced bisulfite-con- verted fragments A and B were investigated [136] : all CpGs found in domain 1 were methylated; all CpGs found in domain 2 also were methylated; in contrast, all CpGs found in domain 3 were unmethylated (starting from site +1, the fragment B, with its domain 3, overlapped a 5’-UTR, also unmethylated). In HUVEC cells, carrying an active hTGC gene, several CpGs of domain 1 were unmethylated [136] [138] .
base-sequence analysis showing the inverse correlation between the activity of a gene and the methylation of its promoter (of course, in harmony with this correlation, the on-going research is aimed at clarifying the regulatory role of m5Cs along the non-coding sequences).
9. Interpretations of the Mechanisms Switching-on and -off Transcription in Higher Cells: Future Perspectives
From the picture described herewith what hypothesis could one deduce on the role of methylation in regulating gene activity?
Supporting the message coming from Figure 8 and Figure 10 and from the long list of genes regulated by methylation [98] [99] , the analysis based on the use of methylation-sensitive restriction endonucleases further showed that in human lymphocytes and monocytes an inverse correlation between the hTGc gene expression and methylation of its promoter took place [135] .
Furthermore, the study based on the direct sequencing of m5C showed that in the same cell species, in the absence of hTGc activity, the hTGc promoter was hypermethylated in its CpG-rich domains 1 and 2 and unmethylated in its CpG-rich domain 3 [136] .
This neat division of hTGc promoter in two parts―one methylated and the other not―deserved particular attention in correlating DNA heterochromatization with A or B m5CpG-binding proteins [123] [139] -[141] [139] -[141] : one could assume that they might transform domains 1 and 2 into heterochromatic structures and that, in turn, these structures would be sufficient to prevent transcription (methylation would be relevant to the repressor complex able to bind the promoter 5’-end).
Then, one could ask why, in hTGc promoter, the unmethylated domain 3, remaining in normal relaxed state, would be unable to interfere with the basal transcription mechanism at the promoter 3’-end [138] .
Combined with the inactivity of the hTGc gene in leukocytes [136] , the postulated heterochromatization of domains 1 and 2, in its promoter, could acquire great interest if considered within the framework of the repair- modification scheme (Figure 6(b)). Causing an at random DNA de-methylation [95] [142] , it would lead, in general, to switch-on of previously silent genes, as in the case of genes for alpha and beta chains of hemoglobin in Friend erythroleukemia cells [99] [143] .
In this situation, the switch-on and -off of transcription―implying conformational changes of the two hemoglobin gene promoters―would correspond, first, to their de-methylation and, second, to their re-methylation [99] [143] [144] .
In other words, following the damages caused to double helix by ionizing radiations [95] [142] , excision-re- pair would be sufficient to guarantee a complete reconstruction of previously unmethylated regions; nevertheless, to complete the reconstruction of previously methylated regions, a coupling would arise [53] between excision- repair (re-establishing the basic code in A, G, T, and C) and DNAmet (re-establishing the position of m5C among A, G, T, and C). Actually, a coupling (properly meaning repair-modification) took place in S [64] : a specific DNApol, crucial for excision-repair, was active during the whole cell cycle [112] ; DNAmet was highly active in S and almost inactive during the major part of the extra-S time [63] . Once de-methylated through excision-repair, given genes, silent when methylated, could be expressed if their transcription was inversely correlated with their methylation.
10. Conclusion
In conclusion, our basic hypothesis was that m5C might function as a signal for genetic regulation of transcription through the hypermethylated promoter and the intermittent methylated non-coding regions. After a possible association with m5C-binding proteins, they would physically interfere with the slip of RNA polymerase (RNApol) on the transcriptional unit. Probably, the block of transcription starts from the methylated promoter (initially recognized by a type A m5C-binding protein able to compete with RNApol) and then is potentiated, inside the gene, by an intermittent association of uncoding methylated regions with a type B m5C-binding protein.
Acknowledgements
The re-visited “pioneering experiment on gene fragmentation” is dedicated to F.H.C. Crick, who inspired me with his enthusiastic lectures on the structure of DNA and the genetic code at the International Institute of Genetics and Biophysics (IIGB) of CNR in Naples in 1964 and 1965, and to S. Ochoa, who affectionately referred to me as his “nephew” at the end of a research seminar on the language of DNA methylation in normal and virus-transformed cells that I took in 1981 at the Roche Institute of Molecular Biology of Nutley, USA. The investigation was encouraged by the geneticist A. Buzzati Traverso, founder of the IIGB in the 60s, who suggested I study nucleic acids in relation to gene expression, and by the biochemist A. Ruffo, director of the IIGB in the early 70s, who identified the significance of the discovery of non-coding eukaryotic gene sequences to the nascent Genetic Engineering. An appreciation is sincerely expressed to T. Eremenko, whose dedication ensured the success of the research, initiated at the IIGB in Naples and continued for many years both at the Albert Einstein College of Medicine in New York and at the Institute of Experimental Medicine of CNR in Rome. The competence of a large number of coworkers is gratefully acknowledged: R. Sawamura and H.J. Strecker, of the Albert Einstein College of Medicine, New York; E.P. Whitehead, of the Department of Biology of the Commission of the European Communities, Bruxelles; M.Y. Timofeeva, of the Institute of Molecular Biology of the USSR Academy of Sciences, Moscow; R. Cocchiara, D. Geraci, A. Giuditta, A. Granieri, T. Menna, F. Morelli, G. Pagano, and E. Scarano, of the IIGB, Naples; E. Alfani, A.L. Crema, D. Delfini, V. De Venezia, G. Hoberholtzer, G. Sapora, and C. Tomasello, of the High Institute of Health, Rome; G. Elia, C. Esposito, P. Iacovacci, T. Parasassi, and G. Ravagnan, of the Institute of Experimental Medicine of CNR, Rome; R.H. Butler, of the Institute of Cell Biology of CNR, Rome; Benedetto A., of the Laboratory of Virology of the United Hospitals, Rome; Palitti F., of the Department of Biochemical Sciences of the “La Sapienza” University of Rome, Rome; O. Cascio, A. Fazzio, and M.G. Sarpietro, of the Institute of Biochemical and Pharmacological Sciences of the University of Catania, Catania; E. Bertoli, T. Cacciamani, M. Centurelli, T. Duranti, A. La Teana, and S. Virgili, of the Institute of Biochemistry of the University of Ancona, Ancona.
Abbreviations
N6-methyladenine (m6A), 5-methylcytosine (m5C), deoxyadenylate (dam), deoxycytidylate (dcm), methionine (Met), [14C]methyl-L-methionine ([14C]Met), S-adenosyl-L-methionine (AdoMet), bromo-deoxyuridine (BrdUrid), DNA polymerase (DNApol), RNA polymerase (RNApol), methyltransferase (met), transglutaminase (hTGc), housekeeping (HK) and tissuespecific (TS) genes, calcitonin (CALC), mitochondrial DNA (mtDNA), culture growth cycle (CGC), Simian virus 40 (SV40), ultraviolet (UV), repair patch (RP).