A Novel One-Pot and Efficient Procedure for Synthesis of New Fused Uracil Derivatives for DNA Binding ()
1. Introduction
The importance of fused pyrimidines, common source for the development of new potential therapeutic agents [1] [2] , is well known.
Fused pyrimidines continue to attract considerable attention because of their great practical usefulness, primarily due to very wide spectrum of biological activities. This is evident especially from publications of regular reviews on the chemistry of systems where the pyrimidine ring is fused to various heterocycles such as purines, quinazolines, pyridopyrimidines, triazolopyrimidines, pyrazolopyrimidines, pyrimidoazepines, furopyrimidines and pyralopyrimidines.
5-Fluorouracil [3] -[5] and methotrexate (MTX) [6] -[8] are the oldest antifolate anticancer drugs [9] , which are widely used as chemotherapeutic drugs. They compete with the normal substrates, folic acid and dihydrofolate, for the active site on the enzyme dihydrofolate reductase (DHFR) [10] -[12] .
Pyrido[2,3-d]pyrimidines possess dihydrofolate reductase inhibiting and antitumour activity [13] . Similarly, in recent years, considerable attention has been focused on the development of new methodology to synthesize many kinds of pyrazolopyrimidine ring [14] . Indeed, pyrazolopyrimidines [15] [16] and purines [17] represent an important class of heterocyclic compounds having wide range of pharmaceutical and biological activities. Therefore, versatile and widely applicable methods for the synthesis pyrazolopyrimidines and purines are of considerable interest. The existing methods for the preparation of pyrazolopyrimidines are based on heterocyclic hydrazones or hydrazine precursors. Pyrimidines and their derivatives are considered to be important for drugs. A large number of pyrimidine derivatives are reported to exhibit antimycobacterial [18] , antitumor [19] , antiviral [20] , anticancer [21] [22] activities. In the present study, a series of new pyrimidine fused ring analogs have been synthesized and their biological effects are determined.
2. Material and Methods
2.1. Chemistry
All melting points were determined with an Electrothermal Mel.-Temp. II apparatus and were uncorrected. Element analyses were performed at the Micro Analytical Unit, Chemistry Department, Mansoura University. The infrared (IR) spectra were recorded using potassium bromide disc technique on Nikolet IR 200 FT IR at Pharmaceutical Analytical Unit, Faculty of Pharmacy, Al-Azhar University. The proton nuclear magnetic resonance (1H-NMR) spectra were recorded on Varian Gemini 300 MHz Spectrometer using DMSO-d6 as a solvent and tetramethylsilane (TMS) as an internal standard (Chemical shift in δ, ppm), Faculty of Science, Chemistry Department, Cairo University. Mass spectra were recorded on DI-50 unit of Shimadzu GC/MS-QP 5050A at the Regional Center for Mycology and Biotechnology at Al-Azhar University. All reactions were monitored by TLC using precoted plastic sheets silica gel (Merck 60 F254) and spots were visualized by irradiation with UV light (254 nm). The used solvent system was chloroform: methanol (9:1) & ethyl acetate: toluene (1:1).
6-(2-Arylidenehydrazin-1-yl)-1-methyluracils (4a-f) [23]
A mixture of 6-hydrazinyl-1-methyluracil (3) (0.4 g, 2.5 mmol) and the appropriate aromatic aldehyde (2.5 mmol) in ethanol (25 ml) was stirred at room temperature for 1.5 - 2 hours. The formed precipitate was filtered, washed with ethanol and crystallized from DMF/ethanol (2:1) into yellow crystals.
Benzaldehyde(3-methyl-2,6-dioxo-1,2,3,6-tetrahydropyrimidin-4-yl)hydrazine 4a [23] : Yield: 81%, m.p. = 276˚C - 277˚C [23] .
4-Methoxybenzaldehyde(3-methyl-2,6-dioxo-1,2,3,6-tetrahydropyrimidin-4-yl)hydrazine 4b [23] : Yield: 94%, m.p. = 266˚C - 268˚C [23] .
4-Hydroxybenzaldehyde(3-methyl-2,6-dioxo-1,2,3,6-tetrahydropyrimidin-4-yl)hydrazine 4c: Yield: 79%, m.p. = 254˚C - 256˚C. IR= 3300-3136 overlapped (OH &NH), 3010 (CH-arom.), 2840 (CH-aliph.), 1706 (2 C = O), 1644 (C = N), 832 (p-substituted phenyl). Anal. Calcd for C12H12N4O3 (260.25), Calcd.: C, 55.38, H, 4.65, N, 21.35, Found: C, 55.42, H, 4.70, N, 21.65.
3-Chlorobenzaldehyde(3-methyl-2,6-dioxo-1,2,3,6-tetrahydropyrimidin-4-yl)hydrazine 4d: Yield: 91%, m.p. = 261˚C - 263˚C. IR= 3252 (NH), 3102 (CH-arom.), 2900 (CH aliph.), 1728 (2 C = O), 1636 (C = N), 700 & 786 (m-substituted phenyl). Anal. Calcd for C12H11ClN4O2 (278.69), Calcd.: C, 51.72, H, 3.98, N, 20.10, Found: C, 51.41, H, 4.45, N, 20.28.
4-Chlorobenzaldehyde(3-methyl-2,6-dioxo-1,2,3,6-tetrahydropyrimidin-4-yl)hydrazine 4e [23] : Yield: 92%, m.p. = 273˚C - 275˚C [23] .
4-Hydroxy-3-methoxybenzaldehyde(3-methyl-2,6-dioxo-1,2,3,6-tetrahydropyrimidin-4-yl) hydrazine 4f: Yield: 82%, m.p. = 250˚C - 252˚C. IR= 3494 (OH), 3180 (NH), 3036 (CH-arom.), 2920 (CH aliph.), 1708 (2 C = O), 1644 (C = N), 820, 760 (substituted phenyl). Anal. Calcd for C13H14N4O4 (290.27), Calcd.: C, 53.79, H, 4.86, N, 19.30, Found: C, 53.60, H, 4.53, N, 19.10.
3-Aryl-7-methyl-1H-pyrazolo[3,4-d]pyrimidine-4,6(5H,7H)-diones (5a-f)
A mixture of the appropriate 6-(2-arylidenehydrazin-1-yl)-1-methyluracil (4a-f) (1.2 mmol) and excess of thionyl chloride (2 ml) was heated under reflux for 5 - 7 minutes. The excess thionyl chloride was evaporated under reduced pressure. An adequate amount of aqueous ammonia solution was added to the residue. The formed precipitate was filtered, washed with ethanol and crystallized from DMF/ethanol (3:1).
7-Methyl-3-phenyl-1H-pyrazolo[3,4-d]pyrimidine-4,6(5H,7H)-dione 5a: Yield: 69%, m.p. = 212˚C - 214˚C. IR = 3172 (NH), 3050 (CH arom.), 2940, 2868 (CH aliph.), 1716, 1672 (2 C = O), 1563 (C = N) & (C = C). 1H-NMR (DMSO-d6) δ ppm: 11.80 (bs, 1H, NH), 10.86 (s, 1H, NH), 8.06 (s, 2H, arom.), 7.48 (s, 3H, arom.), 3.52 (s, 3H, NCH3). Anal. Calcd for C12H10N4O2 (242.23), Calcd.: C, 59.50, H, 4.16, N, 23.13, Found: C, 58.96, H, 4.35, N, 23.01.
3-(4-Methoxyphenyl)-7-methyl-1H-pyrazolo[3,4-d]pyrimidine-4,6(5H,7H)-dione 5b: Yield: 54%, m.p. = 215˚C - 217˚C. IR = 3173 (NH), 3052 (CH arom.), 2943, 2815 (CH aliph.), 1683 (br, 2 C = O), 1563 (C = N) & (C = C), 844 (p-substituted phenyl). MS: m/z (%) = 272 (M+, 2.52), 85 (100). Anal. Calcd for C13H12N4O3 (272.25), Calcd.: C, 57.35, H, 4.44, N, 20.58, Found: C, 57.01, H, 4.44, N, 20.32.
3-(4-Hydroxyphenyl)-7-methyl-1H-pyrazolo[3,4-d]pyrimidine-4,6(5H,7H)-dione 5c: Yield: 51%, m.p. = 210˚C - 212˚C. IR = 3430 (OH), 3174 (NH), 3053 (CH arom.), 2939, 2817 (CH aliph.), 1697 (2 C = O), 1583 (C = N) & (C = C), 840 (p-substituted phenyl). Anal. Calcd for C12H10N4O3 (258.23), Calcd.: C, 55.81, H, 3.90, N, 21.70, Found: C, 55.56, H, 3.79, N, 21.41.
3-(3-Chlorophenyl)-7-methyl-1H-pyrazolo[3,4-d]pyrimidine-4,6(5H,7H)-dione 5d: Yield: 67%, m.p. = 239˚C - 241˚C. IR = 3171 (NH), 3051 (CH arom.), 2920, 2853 (CH aliph.), 1716, 1676 (2 C = O), 1564 (C = N) & (C = C), 756, 668 (m-substituted phenyl). Anal. Calcd for C12H9ClN4O2 (276.67), Calcd.: C, 52.09, H, 3.28, N, 20.25, Found: C, 51.65, H, 3.68, N, 20.40.
3-(4-Chlorophenyl)-7-methyl-1H-pyrazolo[3,4-d]pyrimidine-4,6(5H,7H)-dione 5e: Yield: 72%, m.p. = 195˚C - 197˚C. IR = 3171 (NH), 3050 (CH arom.), 2936, 2854 (CH aliph.), 1713, 1670 (2 C = O), 1565 (C = N) & (C = C), 824 (p-substituted phenyl). Anal. Calcd for C12H9ClN4O2 (276.67), Calcd.: C, 52.09, H, 3.28, N, 20.25, Found: C, 52.09, H, 3.18, N, 20.08.
3-(4-Hydroxy-3-methoxyphenyl)-7-methyl-1H-pyrazolo[3,4-d]pyrimidine-4,6(5H,7H)-dione 5f: Yield: 63%, m.p. = 192˚C - 194˚C. IR = 3426 (OH), 3172 (NH), 3051 (CH arom.), 2930, 2879 (CH aliph.), 1721, 1671 (2 C = O), 1561 (C = C), 843, 665 (substituted phenyl). Anal. Calcd for C13H12N4O4 (288.25), Calcd.: C, 54.17, H, 4.20, N, 19.44, Found: C, 54.52, H, 4.02, N, 19.31.
6-Amino-1-[(2-chlorophenyl)methyl]-5-nitrosouracil (7)
A mixture of 6-amino-1-[(2-chlorophenyl)methyl]uracil (6) (2.0 g, 7.9 mmol) was suspended in water (90 ml) in the presence of glacial acetic acid (0.4 ml) and sodium nitrite (0.54 g, 7.9 mmol) in water (5 ml ) was stirred at room temperature for 1/2 hr. The formed cherry red precipitate was filtered, washed with ethanol and crystallized from ethanol into violet crystals 7, Yield: 95%, m.p. = 235˚C - 237˚C. IR = 3479 (N-OH), 3338, 3251 (NH2 & NH), 3077 (CH arom.), 2979, 2804 (CH aliph.), 1690, 1638 (2 C = O), 751 (o-substituted phenyl). Anal. Calcd for C11H9ClN4O3 (280.66), Calcd.: C, 47.07, H, 3.23, N, 19.96, Found: C, 47.03, H, 3.20, N, 19.72.
8-Aryl-3-[(2-chlorophenyl)methyl]-7-hydroxyxanthines (8a-d)
A mixture of 6-amino-1-[(2-chlorophenyl)methyl]-5-nitrosouracil (7) (0.3 g, 1.06 mmol) and the appropriate N-arylidene aniline (1.06 mmol) in glacial acetic acid (3 ml) was heated under reflux for 8 - 10 hours. After cooling, the formed precipitate was filtered, washed with ethanol and crystallized from DMF/ethanol (2:1) into colourless crystals.
3-(2-Chlorobenzyl)-8-(4-chlorophenyl)-7-hydroxyxanthine (8a): Yield: 86%, m.p. = >330˚C. IR = 3300 - 2900 (br, OH), 3143 (NH), 3042 (CH arom.), 2823 (CH aliph.), 1695 (C = O), 1548 (C = C), 838 (p-substituted phenyl), 747 (o-substituted phenyl). 1H-NMR (DMSO-d6) δ 14.01 (s, 1H, OH, exchangeable), 11.33 (s, 1H, NH, exchangeable), 8.06 - 8.04 (d, 2H, arom.), 7.56 - 7.52 (d, 2H, arom.), 7.49 (d, 1H, arom.), 7.33 - 7.22 (m, 2H, arom.), 7.08 - 7.05 (d, 1H, arom.), 5.24 (s, 2H, NCH2). Anal. Calcd for C18H12Cl2N4O3 (403.21), Calcd.: C, 53.62, H, 3.00, N, 13.89, Found: C, 53.81, H, 3.15, N, 13.80.
8-(4-Bromophenyl)-3-(2-chlorobenzyl)-7-hydroxyxanthine (8b): Yield: 81%, m.p. = >330˚C. IR = 3300 - 2900 (br, OH), 3150 (NH), 3024 (CH arom.), 2940, 2819 (CH aliph.), 1697 (C = O), 1552 (C = C), 835 (p-substituted phenyl), 749 (o-substituted phenyl). Anal. Calcd for C18H12BrClN4O3 (447.66), Calcd.: C, 48.29, H, 2.70 , N, 12.52, Found: C, 48.49, H, 3.20, N, 12.86.
3-(2-Chlorobenzyl)-7-hydroxy-8-(4-nitrophenyl)xanthine 8c: Yield: 89%, m.p. = >330˚C. IR = 3300 - 3000 (br, OH), 3147 (NH), 3025 (CH arom.), 2925, 2850 (CH aliph.), 1693 (C = O), 1559 (C = C), 1520, 1343 (NO2), 859 (p-substituted phenyl), 752 (o-substituted phenyl). Anal. Calcd for C18H12ClN5O5 (413.77), Calcd.: C, 52.25, H, 2.92, N, 16.93, Found: C, 52.40, H, 2.90, N, 17.22.
3-(2-Chlorobenzyl)-8-(4-fluorophenyl)-7-hydroxyxanthine 8d: Yield: 63%, m.p. = >330˚C. IR = 3300 - 3000 (br, OH), 3155 (NH), 3025 (CH arom.), 2923, 2849 (CH aliph.), 1695 (C = O), 1562 (C = C), 843 (p-substituted phenyl), 747 (o-substituted phenyl). 1H-NMR (DMSO-d6) δ 13.98 (s, 1H, OH), 11.28 (s, 1H, NH), 8.08 - 8.04 (m, 2H, arom.), 7.50 - 7.47 (d, 1H, arom.), 7.33 - 7.24 (m, 4H, arom.), 7.04 - 7.02 (d, 1H, arom.), 5.23 (s, 2H, NCH2). MS: m/z (%) = 388 (M++2, 1.8), 386 (M+, 4.53), 125 (100). Anal. Calcd for C18H12ClFN4O3 (386.76), Calcd.: C, 55.90, H, 3.13, N, 14.49, Found: C, 56.06, H, 3.50, N, 14.14.
1-Benzyl[or(2-chlorophenyl)methyl]-4b,9b-dihydroxy-9b,10-dihydroindeno[2′,1′:4,5] pyrrolo[2,3- d]pyrimidine-2,4,5(1H,3H,4bH)-triones (9a,b)
A mixture of the appropriate 6-amino-1-benzyl-[or (2-chlorophenyl)methyl]uracil (6a,b) (1.2 mmol) and ninhydrin (0.2 g, 1.2 mmol) in ethanol (20 ml) was heated under reflux for 1 hour. The formed precipitate on hot was filtered, washed with ethanol and crystallized from ethanol.
1-Benzyl-4b,9b-dihydroxy-9b,10-dihydroindeno[2’,1’:4,5]pyrrolo[2,3-d]pyrimidine-2,4,5 (1H,3H,4bH)-trione 9a: Yield: 68%, m.p. = 270˚C - 272˚C. IR = 3544 - 3000 (br, OH), 3286, 3182 (NH), 3025 (CH arom.), 2922, 2845 (CH aliph.), 1709, 1656 (C = O), 1553 (C = C), 769, 702 (monosubstituted phenyl). 1H-NMR (DMSO-d6) δ 10.39 (s, 1H, NH), 9.37 (s, 1H, NH), 7.85 - 7.80 (m, 2H, arom.), 7.70 - 7.68 (d, 1H, arom.), 7.59 - 7.57 (d, 1H, arom.), 7.29 - 7.27 (m, 3H, arom.), 7.18 - 7.16 (m, 2H, arom.), 6.81 (s, 1H, OH), 5.98 (s, 1H, OH), 4.94 - 4.90 (d, 1H, NCH2), 4.80 - 4.67 (d, 1H, NCH2). Anal. Calcd for C20H15N3O5 (377.35) Calcd.: C, 63.66, H, 4.01, N, 11.14, Found: C, 63.46, H, 4.10, N, 10.82.
1-(2-Chlorophenyl)methyl-4b,9b-dihydroxy-9b,10-dihydroindeno[2′,1′:4,5]pyrrolo[2,3-d]pyrimidine- 2,4,5(1H,3H,4bH)-trione 9b: Yield: 71%, m.p. = 272˚C - 273˚C. IR = 3600 - 2900 (br, OH), 3286, 3182 (NH), 3025 (CH arom.), 2844 (CH aliph.), 1712, 1661 (C = O), 1560 (C = C), 762 (o-substituted phenyl). 1H-NMR (DMSO-d6) δ 10.48 (s, 1H, NH, exchangeable), 9.41 (s, 1H, NH, exchangeable), 7.79 - 7.77 (d, 2H, arom.), 7.72 - 7.69 (d, 1H, arom.), 7.60 - 7.49 (m, 2H, arom.), 7.35 - 7.21 (m, 2H, arom.), 6.83 - 6.80 (d, 1H, arom.), 6.78 (s, 1H, OH, exchangeable), 5.97 (s, 1H, OH, exchangeable), 5.04 - 4.98 (d, 1H, NCH2), 4.81 - 4.75 (d, 1H, NCH2). MS: m/z (%) = 414 (M++2, 0.2), 412 (M+, 0.47), [395 (1.2), 393 (3.25, M+−H2O)], 44 (100). Anal. Calcd for C20H14ClN3O5 (411.79) Calcd.: C, 58.33, H, 3.43, N, 10.20, Found: C, 58.31, H, 3.42, N, 9.87.
1,3-Dimethyl-2H-indeno[2,1-g]pteridine-2,4,6-(1H,3H)-trione (11)
Two methods were applied for the synthesis of 11:
A) A mixture of 5,6-diamino-1,3-dimethyluracil hydrochloride (10) (0.2 g, 1.00 mmol) and ninhydrin (0.18 g, 1.00 mmol) in ethanol (10 ml) and drops of TEA was added to adjust pH = 8. The reaction mixture was stirred at room temperature for 30 minutes. The formed precipitate was filtered, washed with ethanol and crystallized from ethanol into yellow crystals.
B) A mixture of 5,6-diamino-1,3-dimethyluracil hydrochloride (10) (0.2 g, 1.00 mmol) and ninhydrin (0.18 g, 1.00 mmol) in water (15 ml) and few drops of ammonium hydroxide solution was added to adjust pH = 8 was stirred at room temperature for 1 hour. The formed precipitate was filtered, washed with ethanol and crystallized from ethanol.
Yield: A 60.7%, B 59%, m.p. = >320˚C. IR = 3066 (CH arom.), 2934, 2870 (CH aliph.), 1723, 1672 (C = O), 1567 (C = N), 1508 (C = C). 1H-NMR (DMSO-d6) δ 7.97 - 7.95 (d, 1H, arom.), 7.85 - 7.81 (m, 2H, arom.), 7.74 - 7.69 (d, 1H, arom.), 3.66 (s, 3H, NCH3), 3.41 (s, 3H, NCH3). MS: m/z (%) = 294 (M+, 100). Anal. Calcd for C15H10N4O3 (294.26) Calcd.: C, 61.22, H, 3.43, N, 19.04, Found: C, 61.03, H, 3.06, N, 18.60.
2.2. Biological Evaluation
2.2.1. Nucleic Acids Preparation
For extraction of genomic DNA, yeast cells were washed with cold phosphate borate sodium chloride (PBS) buffer and lysed in a buffer containing 50 mM Tris-HCl (pH 8.0), 1 mM EDTA, 0.2% Triton X-100 for 20 min at 4˚C. After centrifugation at 14,000 rpm for 15 min, the supernatant was treated with proteinase K (0.5 mg/ml) and 1% SDS for 1 h at 50˚C. DNA was extracted twice with buffered phenol/chloroform and precipitated with 140 mM NaCl and 2 volumes of ethanol at −20˚C overnight. DNA precipitates were washed twice with 70% ethanol, air-dried and dissolved in TE buffer, and treated for 1 h at 37˚C with RNase A according to reported method [24] . Finally, DNA preparations were electrophoresed in 1% agarose gels.
2.2.2. Agrose Gel Preparation and Visualization of DNA
1% agarose gel was prepared by adding 1 gm ultra agarose to 100 ml Tris-Acetate-EDTA (TAE) buffer and heated in a microwave oven then cooled to ~60˚C before pouring in gel tray.
Examination of the gel was carried out using ultraviolet illuminated box. Ethidium bromide (0.1 mg/ml) solution was used to stain the nucleic acid (DNA bands) in the gel as it intercalates between DNA bases and give florescence. The gel was photographed using polarized camera.
2.2.3. Nucleic Acid Affinity, Binding and Fragmentation Assay
The test compounds were dissolved in DMSO at 20 µg/µl concentrations, mixed with 2 µg/µl
DNA
and incubated at room temperature for 2 hrs. The mixtures were mixed with the gel loading buffer and then electrophoresed in the agarose gel (1% w/v) at 80 V for 1.5 hrs. As positive control for affinity, binding and fragmentation, methotrexate (20 µg/µl) was mixed with
DNA ,
and as negative control DMSO was mixed with equal amount of
DNA
. After running, agarose gels were stained with ethidium bromide and visualized using polarized camera.
3. Results and Discussion
3.1. Chemistry
6-Chlorouracils (1) were prepared by the alkaline hydrolysis of 2,4,6-trichloropyrimidines [25] [26] . Methylation of 6-chlorouracil (1) was carried out with methyl iodide in the presence of potassium carbonate applying a reported procedure [27] . 6-Hydrazinyl-1-methyluracil (3) [28] was prepared in a good yield by the reaction of 6-chloro-1-methyluracil (2) with alcoholic hydrazine hydrate at room temperature following a reported method [23] . In this investigation the title compounds were furnished through the hydrazinolysis of 6-chloro-1-methy- luracil (2). Condensation of the hydrazinylpyrimidine 3 with aromatic aldehydes gave the respective hydrazones 4a-f. Oxidative cyclization of 4 using thionyl chloride produced pyrazolopyrimidines 5a-f in good yields.

Thus, refluxing of compounds 4a-f with thionyl chloride resulted in intramolecular cyclization affording pyrazolopyrimidines 5a-f presumably via the formation of the 5-chlorosulfinyl derivatives A which loses (SO) group and HCl to form Xa-f. The structure of target compounds was confirmed by element analysis in addition to IR, 1H-NMR spectral data.
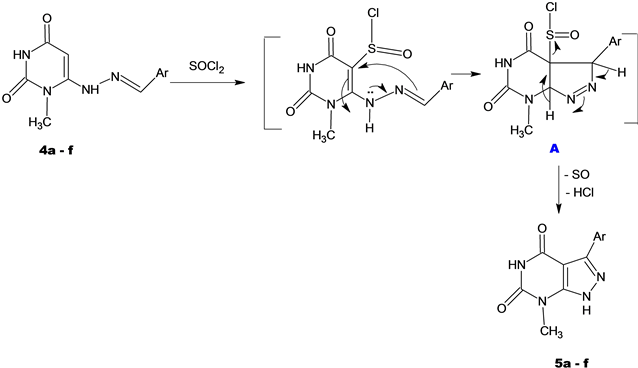
Compounds 6a,b were prepared in good yields by the condensation of ethyl cyanoacetate with N-benzyluea [29] or N-[(2-chlorophenyl)methyl]urea [30] in sodium ethoxide or methoxide. In this work, it was in need to prepare first the unavailable starting material, 6-amino-1-[(2-chlorophenyl)methyl]-5-nitrosouracil (7). Reaction of 6-amino-1-[(2-chlorophenyl)methyl]uracil (6b) with aqueous sodium nitrite in the presence of acetic acid afforded a high yield of the coloured nitroso derivative 7 [31] . Thus, reaction of 7 with different arylidene aniline in acetic acid took place by the elimination of aniline to give 8a-d.

An expected mechanism might be as follows:

On the other hand, the reaction of aminouracils 6a,b by refluxing with ninhydrin in DMF resulted in the formation of indenopyrrolopyrimidines 9a,b in a moderate yields. It was reported that the 2-position of the ninhydrin is more reactive towards nitrogen [32] , oxygen [32] [33] and carbon based nucleophiles [32] -[34] . The cyclization affording 9a,b presumably occurred via the formation of nonisolable acyclic intermediate. The latter might be formed via the attack of the more nucleophilic carbon at 5-position of uracil to the more reactive center at 2-position of ninhydrin. Cyclization could be affected via the addition of the amino group to the carbonyl at 1-position of ninhydrin moiety affording the final product 9a,b. 1H-NMR showed the two benzylic hydrogens as two doublets at δ = 4.94 - 4.67 ppm which indicated that they were not magnetically equivalent. This observation may be attributed to the presence of stereoisomers resulted from the two asymmetric carbons at 4b and 9b positions.

The project now directed towards the possible utility of diaminouracils for the synthesis of the title compound 11. Thus, the reaction of 5,6-diamino-1,3-dimethyluracil hydrochloride (10) [29] [35] -[37] with ninhydrin in the presence of triethylamine or ammonium hydroxide afforded the title compound 11.
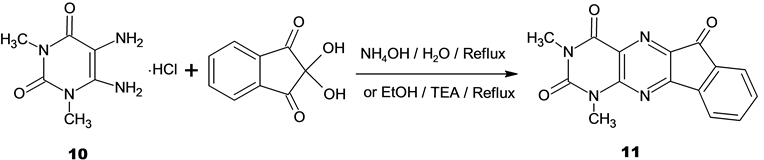
The formation of 11 from the aminouracil 10 and ninhydrin may be proceed through first condensation between the more reactive NH2 at 5-position with the more electrophilic center at C-2 of ninhydrin. Attack of the less reactive NH2 group at 6-position to one of the C = O groups of the reagent afforded the cyclized tautomer B which was stabilized by loss of H2O to give 11.

![]()
Figure 1. Gel electrophoresis 1% w/v agrose of untreated and treated DNA. Lane M: Molecular weight marker (left side); Lane 1: Untreated nucleic acid; Lane 2: DMSO treated nucleic acid (negative control); Lane 3: Methotrexate treated nucleic acid (positive control); Lanes 4-17: Compounds (8a-d, 7, 9a, b, 11, 5a-f) treated nucleic acid.
3.2. Biological Evaluation
The newly synthesized compounds were subjected to nucleic acid binding assay using agarose gel electrophoresis method.
Nucleic Acids Binding Assay
Different synthetic drugs induced DNA damage was evaluated by measuring the level of genomic DNA fragmentation and detecting DNA ladders on agarose gel electrophoresis (Figure 1). Compared with the vehicle control group (lane 2 negative control and lane 3 positive control), there was no significant change in genomic DNA fragmentation in some treated groups. There were major differences in the response of extracted DNA (from Lanes 4-17 in Figure 1). It is possible that drugs exert its effect solely by indirect mechanisms. This contrast may have been due to different enzyme(s) being with differing susceptibilities to drugs.
4. Conclusion
Our results describe a simple and efficient method for the synthesis of different novel fused uracils. Heteroannulation on the C-5 of uracil usually requires forcing conditions and complex synthetic pathways. Our synthetic compounds concern with the reactions of uracils with different benzylideneaniline, araldehydes and ninhydrin which have a biological screen.
Acknowledgements
The authors wish to thank Dr. Yassin El-Ayouty, professor of Microbiology, Botany and Microbiology Department, Faculty of Science, Zagazig University, Zagazig, Egypt, for carrying out the biological activities of the new synthesized compounds.