Synthesis of a Rhodamine-Appended Cyclophane as a Fluorescence Host in Water ()
1. Introduction
In recent years, much attention has been focused on development of fluorescent host sensor systems, which are able to detect small organic compounds [1] . Many types of fluorophore-appended macrocyclic hosts based on cyclodextrins [2] , calixarenes [3] , and cyclophanes [4] were widely investigated. Numerous successful studies of fluorophore-appended hosts based on these macrocyclic compounds were reported [5] . Among them, azacyclophanes [6] having a hydrophobic internal cavity are favorable candidates as the framework of macrocyclic host, because shape and size of the cavity can be easily designed for the capture of target guest molecules. In addition, exterior modifications of azacyclophanes can be achieved by the introduction of various functional groups such as polar side chains for water-solubility and fluorophores for fluorescent sensing onto the nitrogen atoms through an appropriate spacer [7] . In the preceding paper, we have developed water-soluble blue fluorescent cyclophanes [8] , which are composed of a tetraaza[6.1.6.1]paracyclophane skeleton, three polar side chains for water-solu- bility, and a pyrene fluorophore. The pyrene-appended cyclophanes showed characteristic fluorescence spectra originated from pyrene moiety in aqueous media upon irradiation with UV light [8] . A fluorescence intensity originated from the pyrene-appended host decreased upon addition of 8-anilino-naphthalene-1-sulfonate (ANS) as a guest [9] , reflecting the formation of host-guest complexes [8] .
On the other hand, many types of fluorescent dyes such as fluorescein isothiocyanate [10] , rhodamine derivatives [11] , and molecular beacons [12] have been designed and developed in order to investigate interactions of biomolecular complexes and assemblies. Among them, rhodamine derivatives emitting in the red region of visible spectrum are widely used as fluorescent labels for lipids, proteins, peptides, nucleic acids, and other biomolecules [13] . They display high absorption coefficients and emission in the visible region, high fluorescence quantum yields, and high chemical stability and photostability [11] . In the course of our ongoing research on cyclophanes capable of performing guest-inclusion and fluorescent sensing, we became interested in developing fluorescent cyclophanes emitting in longer wavelength ranges than UV waves. As a water-soluble red fluorescent cyclophane, we have now designed cationic cyclophane bearing a rhodamine moiety (1a) and analogous anionic cyclophane (1b) (Figure 1). We report here the synthesis of water-soluble cyclophane having a rhodamine moiety and its guest-binding abilities.
2. Experimental Section
2.1. General Methods
HEPES (N-(2-hydroxyethyl) piperazine-N’-2-ethanesulfonic acid) buffer (0.01 M, pH 7.4, with 0.15 M NaCl) was purchased from GE Healthcare. A cyclophane derivative bearing N-protected amines (2) was prepared after a method reported previously [14] . Elemental analyses were recorded on a Yanako CHN Corder MT-5. 1H and 13C spectra were taken on Varian Mercury 400 spectrometer. Fluorescence spectra, IR spectra, and ESI TOF MS
![]()
Figure 1. Cationic and anionic cyclophanes bearing a rhodamine moiety 1a and 1b.
were recorded on JASCO FP-750, Perkin-Elmer spectrum one, and JMS-T100 CS spectrometers, respectively.
2.2. Precursor of 1a (3)
Piperidine (1.0 mL) was added to a solution of cyclophane derivative bearing N-protected amines (2) (179 mg, 0.14 mmol) in dry dichloromethane (DCM, 5 mL), and the mixture was stirred for 5 h at room temperature. Then the solvent was evaporated off under reduced pressure to give a pale yellow solid (monoamine of cyclophane). The monoamine of cyclophane was purified by gel filtration chromatography on a column of Sephadex LH-20 with methanol as an eluant. The precursor fraction was evaporated to dryness under reduced pressure to give a pale yellow solid (cyclophane monoamine, 152 mg). Triethylamine was added to a solution of the monoamine of cyclophane (140 mg, 0.13 mmol) in dry DCM (8 mL) at room temperature, and the mixture was allowed to stand at same temperature. The mixture was added to a solution of rhodamine B isothiocyanate (91 mg, 0.17 mmol) in dry DCM (2 mL), and the resulting mixture was stirred for 1 day at the same temperature. After being dried (Na2SO4), the solution was evaporated to dryness under reduced pressure to give a dark purple solid. The crude product was purified by gel filtration chromatography on a column of Sephadex LH-20 with methanol as an eluant. Evaporation of the product fraction under reduced pressure gave a dark purple solid (151 mg, 73%): mp 144˚C - 145˚C. 1H NMR (400 MHz, CDCl3, 293 K) δ 1.3 (m, 12H), 1.4 (m, 35H), 2.1 (m, 8H), 3.3 (m, 8H), 3.5 (m, 8H), 3.6 (m, 8H), 3.9 (m, 4H), 5.3 (m, 5H), 6.6 (m, 4H), 7.0 (m, 10H), 7.1 (m, 10H) and 7.5 (m, 1H). 13C NMR (100 MHz, CDCl3, 293 K) δ 12.8, 25.1, 28.6, 35.0, 36.5, 40.3 - 41.5, 45.8, 48.9, 79.1, 96.2 - 96.9, 112 - 113, 128 - 129, 130 - 131, 132, 140 - 141, 155, 156, 157, 171, 172 and 181. IR 1646 cm−1 (C=O). Found: C, 59.72; H, 7.47; N, 8.74. Calcd for C90H113ClN11NaO13 S・ 9H2O: C, 59.74; H, 7.30; N, 8.51. ESI-TOF MS (positive mode): m/z 1589 [M + H]+, 1611 [M + Na]+, where M denotes zwitterionic form of cyclophane (M, C90H113N11 O13S).
2.3. Cationic Cyclophane Bearing a Rhodamine Moiety (1a)
Trifluoroacetic acid (1.0 mL) was added to a solution of CP-Boc3RhB (153 mg, 0.096 mmol) in dry DCM (6 mL), and the mixture was stirred for 1 h at room temperature. Evaporation of the solvent under reduced pressure gave a dark purple solid. The crude product was purified by gel filtration chromatography on a column of Sephadex LH-20 with methanol as an eluant. Evaporation of the product fraction under reduced pressure gave a dark purple solid (139 mg, 89%): mp 182˚C - 190˚C (decomp.). 1H NMR (400 MHz, CD3OD, 293 K) δ 1.1 - 1.2 (m, 12H), 1.3 - 1.5 (m, 8H), 2.2 - 2.5 (m, 8H), 2.9 - 3.1 (m, 8H), 3.5 - 3.8 (m, 16H), 3.9 - 4.1 (m, 4H), 6.7 - 7.0 (m, 8H), 7.1 - 7.4 (m, 16H) and 8.1 (m, 1H). 13C NMR (100 MHz, CD3OD, 293K) δ 11.8, 23.6 - 25.0, 31.4, 35.8, 40.6, 45.5, 48.6, 95.8, 113 - 114, 116 - 121, 128 - 129, 130 - 131, 132, 139 - 141, 142, 155, 158, 161 - 162, 170 and 181. IR 1645 cm−1 (C=O). Found: C, 58.52; H, 6.00; N, 9.28. Calcd for C81H92F9N11O13S・2H2O: C, 58.37; H, 5.81; N, 9.24. ESI-TOF MS (positive mode): m/z 1289 [M + H]+, 1311 [M + Na]+, where M denotes triamine derivative of cyclophane as a free base (M, C75H89N11O7S).
2.4. Anionic Cyclophane Bearing a Rhodamine Moiety (1b)
Succinic anhydride (59 mg, 0.59 mmol) was added to a solution of cyclophane 1a (106 mg, 0.06 mmol) and triethylamine (0.5 mL) in dry DCM (4 mL) at room temperature, and the mixture was stirred for 1 day. Ethylenediamine (0.1 mL, 1.5 mmol) was added to the mixture to quench the reaction. After being dried (Na2SO4), the solution was evaporated to dryness under reduced pressure to give a dark purple solid. The crude product was purified by gel filtration chromatography on a column of Sephadex LH-20 with methanol as an eluant. Evaporation of the product fraction under reduced pressure gave a dark purple solid. Then added 0.1 M NaOH aq. (2 ml) and stirred 20 min at room temperature. After dialysis (1.0 kDa cut-off) for 4 h, the solvent was freeze-dried to gave a dark purple solid (88 mg, 82 %): mp 170˚C - 172˚C (decomp.). 1H NMR (400 MHz, CD3OD, 293 K) δ 1.0 - 1.5 (m, 20H) 2.2 (m, 6H), 2.4 (m, 14H), 3.3 (m, 8H), 3.4 - 3.8 (m, 16H), 3.9 - 4.0 (m, 4H), 6.8 (m, 4H), 6.9 - 7.1 (m, 10H), 7.2 - 7.4 (m, 8H), 7.7 - 7.9 (m, 2H) and 8.1 (m, 1H). 13C NMR (100 MHz, CD3OD, 293K) δ 11.7, 23.1, 23.9, 32.8, 33.5, 34.1, 35.4, 40.6, 45.6, 52.0, 96.0, 113, 114, 128 - 129, 130 - 131, 132, 140, 141 - 142, 155, 158, 171 - 172, 174 ,179, 180 and 181. IR 1736, 1635 cm−1 (C=O). Found: C, 63.37; H, 6.35; N, 9.56. Calcd for C87H101N11O16S・3H2O: C, 63.60; H, 6.56; N, 9.38. ESI-TOF MS (negative mode): m/z 1589 [M − H]−, 1610 [M − 2H + Na]−, 1632 [M − 3H + 2Na]−, where M denotes carboxylic acid of cyclophane (M, C87H101N11O16S).
2.5. C-Protected Dabsyl Guest (4)
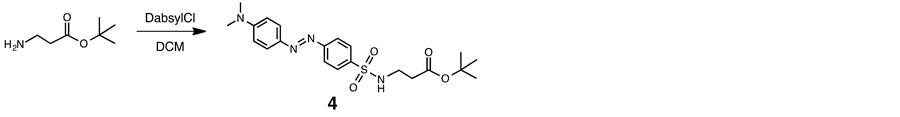
Triethylamine (0.5 mL) was added to a solution of β-alanine t-butyl ester hydrochloride (95 mg, 0.52 mmol) in dry DCM (10 ml) at room temperature. The mixture was added to a solution of 4-dimethyl-aminoazobenzene- 4-sulfonyl chloride (DabsylCl, 149 mg, 0.46 mmol) in dry DCM (5 ml), and the resulting mixture was stirred for day at room temperature. The residue was chromatographed on a column of silica gel (SiO2) with chloroform-methanol (95:5 v/v) as eluant. Evaporation of the product fraction under reduced pressure gave a orange-red solid (158 mg, 79%): mp 170˚C - 171˚C. 1H NMR (400 MHz, CDCl3, 293 K) δ 1.4 (s, 9H), 2.4 (m, 2H), 3.2 (m, 6H), 3.5 (m, 2H), 6.8 (m, 2H) and 7.9 - 8.0 (m, 6H). 13C NMR (100 MHz, CDCl3, 293K) δ 28.3, 35.1, 39.2, 40.5, 81.8, 112, 122, 126, 140, 144, 153, 156 and 172. IR 1708 cm−1 (C=O). Found: C, 57.11; H, 6.38; N, 12.63. Calcd for C21H28N4O4S∙0.5 H2O: C, 57.12; H, 6.62; N, 12.69. ESI-TOF MS (positive mode): m/z 433 [M + H]+, 455 [M + Na ]+.
2.6. Anionic Dabsyl Guest (5)
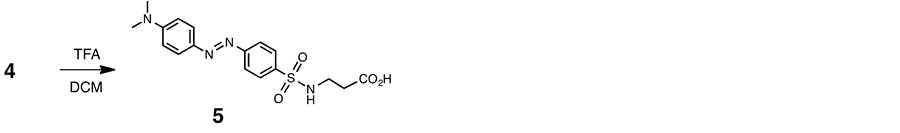
Trifluoroacetic acid (1.0 ml) was added to a solution of 4 (75 mg, 0.17 mmol) in dry DCM (5 ml), and the mixture was stirred for 4 h at room temperature. The residue was chromatographed on a column of silica gel (SiO2) with chloroform-methanol (9:1 v/v) as eluant. The product fraction was evaporated to dryness under reduced pressure to give a orange-red solid (53 mg, 82%): mp 164˚C - 165˚C. 1H NMR (400 MHz, CD3OD, 293 K) δ 2.5 (m, 2H), 3.1 (m, 6H), 3.2 (m, 2H), 6.9 (m, 2H) and 7.9 - 8.0 (m, 6H). 13C NMR (100 MHz, CD3OD, 293K) δ 28.3, 35.1, 39.2, 40.5, 81.8, 112, 122, 126, 140, 144, 153, 156 and 172. IR 1709 cm−1 (C=O). Found: C, 54.24; H, 5.36; N, 14.88. Calcd for C17H20N4O4S: C, 54.00; H, 5.40; N, 15.11. ESI-TOF MS (positive mode): m/z 377 [M + H]+, 399 [M + Na ]+.
2.7. Computational Procedure
The calculations were carried out on a Pentium 4 3.2 GHz × 2 machine using Macro Model 9.1 molecular modeling software on a Red Hat Enterprise Linux WS 4.3 operating system. The geometry of 1a and 1b was optimized using molecular mechanics employing the OPLS_2005 force field for the simulation of the hosts. The geometry was optimized without any constraints allowing all atoms, bonds, and dihedral angles to change simultaneously.
2.8. Binding Constants of Cyclophanes with the Guests
To each solution of fluorescent cyclophane (0.5 μM) in HEPES buffer were added increasing amounts of 5 and 6, and the fluorescence intensity was monitored after each addition by excitation at 558 nm. Aqueous stock solution of 5 was prepared after addition of NaOH. The binding constants were calculated on the basis of the Benesi-Hildebrand method for titration data.
3. Results and Discussion
3.1. Design and Synthesis of Rhodamine-Appended Cyclophanes
From a viewpoint of development of cyclophanes emitting in the red region of visible spectrum, we have designed water-soluble cyclophanes having a rhodamine moiety. Actually, we have adopted a simple strategy to prepare rhodamine-appended cyclophanes by introducing a rhodamine moiety into tetraaza[6.1.6.1]paracyclo- phane [15] through a β-alanine spacer. Rhodamine-appended cyclophanes bearing cationic and anionic polar side chains 1a and 1b, respectively, were synthesized by following the reaction sequence shown in Scheme 1. In the preceding paper, we have synthesized a cyclophane derivative bearing N-protected amines 2 as a key intermediate [14] . A precursor (3) of 1a was synthesized by a reaction of rhodamine B isothiocyanate (RITC) [16] with a monoamine derivative of cyclophane, which was easily prepared from 2 by removal of the Fmoc protecting group with piperidine, in a 73% yield. Cationic cyclophane bearing a rhodamine moiety 1a was derived from 3 by a treatment with trifluoroacetic acid (TFA). Then, 1a was converted to a cyclophane having carboxylic acid residues 1b by a reaction with succinic anhydride. New compounds were fully characterized by means of spectroscopy (1H and 13C NMR, and TOF-MS) and elemental analysis. Even though compounds 1a and 1b contain a hydrophobic cavity, both compounds were soluble in aqueous neutral media at biological pH owing to three polar side chains. From a practical standpoint, cyclophanes 1a and 1b had good H2O-solubility of 0.27 and 0.38 g/mL, respectively. Judging from molecular mechanics studies of cyclophanes 1a and 1b, both compounds provide a rigid internal cavity and the peripheral polar side chains with reasonably separated distances from the cavity (Figure 2). These results indicate that 1a and 1b having hydrophobic cavities were expected to act as water-soluble hosts.
3.2. Guest-Binding Behavior of Cyclophanes
As mentioned above, rhodamine derivatives have an intense visible absorption. Actually, rhodamine-appended water-soluble cyclophanes 1a and 1b had high absorption coefficients and absorption in the visible region owing to the rhodamine moieties. In addition, they showed fluorescence emission spectra originated rhodamine moieties with a fluorescence maximum at 579 nm in aqueous media in aqueous HEPES (2-[4-(2-hydroxy- ethyl)-1-piperazinyl]ethanesulfonic acid) buffer (0.01 M, pH 7.4, 0.15 M with NaCl) at 298 K (Figure 3 for 1a). First, the guest-binding behavior of 1a toward anionic dabsyl derivative 5 as a dark quencher guest, was examined by fluorescence spectroscopy in aqueous HEPES buffer (0.01 M, pH 7.4, 0.15 M with NaCl). The fluorescence intensity originated from 1a at 579 nm decreased upon addition of 5, reflecting formation of 1a•5 complexes, as shown in Figure 3(a). The stoichiometry for the complex was confirmed to be 1:1 1a:5 by a Job plot (Figure 4(a)). The 1:1 binding constant (K) of 1a toward 5 was calculated to be 2.7 × 104 M−1 on the basis of the Benesi-Hildebrand relationship. On the other hand, the K value of anionic cyclophane 1b with the identical guest 5 was not determined due to the low affinity in HEPES buffer by the identical method. These results indicate that the electrostatic interaction between host and guest molecules is effective recognition factor for the
![]()
Scheme 1. Preparation of rhodamine-appended cyclophanes 1a and 1b.
![]()
Figure 2. Computer-generated CPK models for 1a (a) and 1b (b). Carbon, hydrogen, oxygen, nitrogen, and sulfur atoms are shown in green, white, red, blue, and yellow respectively.
![]()
![]()
Figure 3. Fluorescence spectral changes for complexes of 1a with 5(a) and 6(b) in HEPES buffer (0.01 M, pH 7.4, 0.15 M with NaCL) at 298 K. [1a] = 0.5 μM. [5] = [6] = 0, 5, 10, 15, 20, 25, 30, 35, 40, 45, and 50 μM. (from top to bottom). Ex. 558 nm. Insets: the corresponding titration curves.
host-guest complexation. A similar fluorescence feature was observed when 4-(1-pyrene)butanoate (6) was employed as an anionic florescence guest. That is, upon addition of 6 to an aqueous solution containing 1a, fluorescence intensity originated from 1a decreased, as shown in Figure 3(b), reflecting the formation of host-guest complexes. Such fluorescence quenching of 1a at 579 nm seems to be caused by the interactions between rhodamine group of 1a and entrapped pyrene molecule. The stoichiometry for the complex was also confirmed to be 1:1 1a:6 by a Job plot (Figure 4(b)). The K value of 1a with 6 was calculated to be 2.9 × 104 M−1, which was almost comparable to that of 1a with 5.
4. Conclusion
Rhodamine-appended cyclophanes bearing three cationic polar side chains 1a were successfully prepared by
![]()
Figure 4. Job’s plots for complex of 1a and 5(a), 1a and 6(b): [1a] + [5] = [1a] + [6] = 1.0 μM.
reaction of RITC with a monoamine derivative of cyclophane, followed by removal of the protecting groups in a fairly good yield. 1a showed fluorescence bands with a fluorescence maximum at 579 nm in an aqueous HEPES buffer. Formation of the host-guest complexes of the present cyclophane with anionic guests was demonstrated by fluorescence quenching experiments. The fluorescence intensity originating from 1a was subjected to decrease, upon complexation with anionic guests such as 5 and 6.
Acknowledgements
The present work is partially supported by Grant-in-Aid (No. 24550166) from the Ministry of Education, Culture, Science, Sports and Technology of Japan.
NOTES
*Corresponding author.