Familial Hypercholesterolemia in an Azorean Family: A Novel Mutation in the Low-Density Lipoprotein Receptor Gene ()
1. Introduction
Heterozygous familial hypercholesterolemia (FH) is the most common inherited form of primary hyperlipidemia, with an incidence around 1:500 newborns. The disease is still under-diagnosed in many populations, including the Portuguese [1] . The total number of FH patients in Portugal has been estimated to be around 20,000 cases [2] .
FH has a co-dominant transmission and is caused by mutation of low-density lipoprotein receptor (LDLR) gene in most cases. The LDLR binds low-density lipoprotein cholesterol (LDL-C) with high affinity at the hepatic cellular membrane, and then the receptor-ligand complex is internalized and delivered to the endosomes where the LDL-C is released and metabolized. Reduced receptor function (2% - 25% in heterozygotes FH) results in elevated LDL-C levels in blood [3] [4] . There is evidence that the type of LDLR mutation can be the prediction of LDL-C levels and the risk of premature coronary artery disease occurring early in life [5] .
Over the years, other genes associated with the FH phenotype have been identified, such as apolipoprotein B (APOB) and more recently proprotein convertase subtilisin/kexin type 9 (PCSK9) [4] . However, the clinical manifestations of familial defective ApoB are generally milder than those caused by defects in LDLR gene [6] [7] .
In children, FH is diagnosed based on high levels of LDL-C, above the 95th percentile for sex and age together with normal levels of high density lipoprotein cholesterol (HDL-C) and triglycerides, and a family history of hypercholesterolemia and premature CD. When a pediatric patient has LDL-C exceeding 140 mg/dL, a family member(s) diagnosed with FH, and a family history of hyper LDL-C or premature cardiovascular disease (CD), extensive examination must be performed to make a definitive diagnosis [8] .
Patients with FH have increased levels of LDL-C since birth, which left untreated lead to premature CD. In fact, a significant increase in the carotid intima-media thickness is already found at the age of 12-years [9] [10] , reflecting the accelerated development of atherosclerosis in affected patients, which starts much earlier than in hyperlipoproteinemic patients without a genetic trait. This finding reinforces the need for an early diagnosis of FH [9] [11] [12] .
The Portuguese Familial Hypercholesterolemia Study was begun at the National Institute of Health in 1999 [2] . The aim of this study was to perform an epidemiological study to determine the prevalence and distribution of FH in Portugal and to better understand the pathophysiology of coronary heart disease in the patients. The clinical criteria used for the diagnosis of FH were adapted from those of the Simon Broome Heart Research Trust. Genetic diagnosis is based on the search for mutations in three genes: LDLR, APOB and PCSK9.
Within the framework of this epidemiological Study, we present an Azorean family affected by a novel mutation of the LDLR across three generations.
2. Case Report
A 12-year-old boy was first seen at our outpatient consultation because of delayed growth and development. Laboratorial investigations revealed a complete failure of the anterior hypophysis due to a congenital malformation of the sella turcica (Figure 1).
On routine blood tests he presented hypercholesterolemia with total cholesterol of 313 mg/dL (90 - 190 mg/dL); LDL-C 262 mg/dL (<115 mg/dL); HDL-C 43 mg/dL (>40 mg/dL) and triglycerides 42 mg/dL (<150 mg/dL). He was started on hormonal replacement treatment (growth hormone, levothyroxine and hydrocortisone). Initially the high level of cholesterol was thought to be caused by central hypothyroidism. However, making the patient’s thyroid hormone levels into normal status, total cholesterol remained above 300 mg/dL (90 - 190 mg/dL), raising the suspicion of FH.
There was a familial history of CD and premature death (Figure 2).
His father had a myocardial infarction at age 34-years, when cholesterol levels above 500 mg/dL (90 - 190 mg/dL) were firstly noticed. His grandfather died early from myocardial infarction. Hypercholesterolemia was present in three generations: index patient, his father and grandmother. All family members had normal Body Mass Index (BMI).
Given his personal and family history of dyslipidemia and premature coronary disease, the patient was enrolled in the Portuguese FH Study as index-case of his family. His lipid profile at the beginning of the study was: total cholesterol, 290 mg/dL (90 - 190 mg/dL); LDL-C, 237 mg/dL (<115 mg/dL); HDL-C, 26 mg/dL (>40 mg/dL); apoAI, 86 mg/dL (>160 mg/dL); apoB 189 mg/dL (<150 mg/dL); Lp(a) 8 mg/dL (<30 mg/dL); and triglycerides 82 mg/dL (<150 mg/dL).
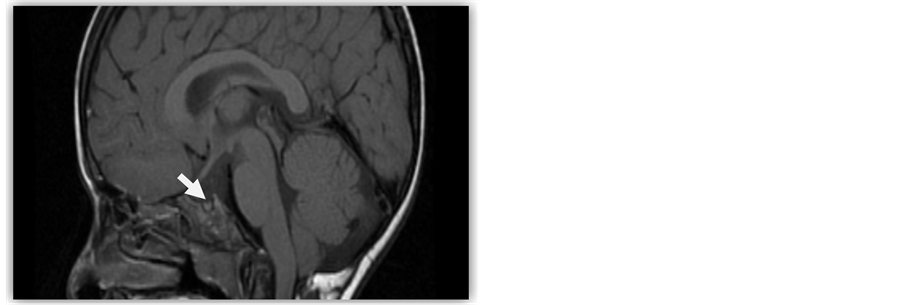
Figure 1. Brain CT scan of the patient with atrophic pituitary gland within the sellar pouch.
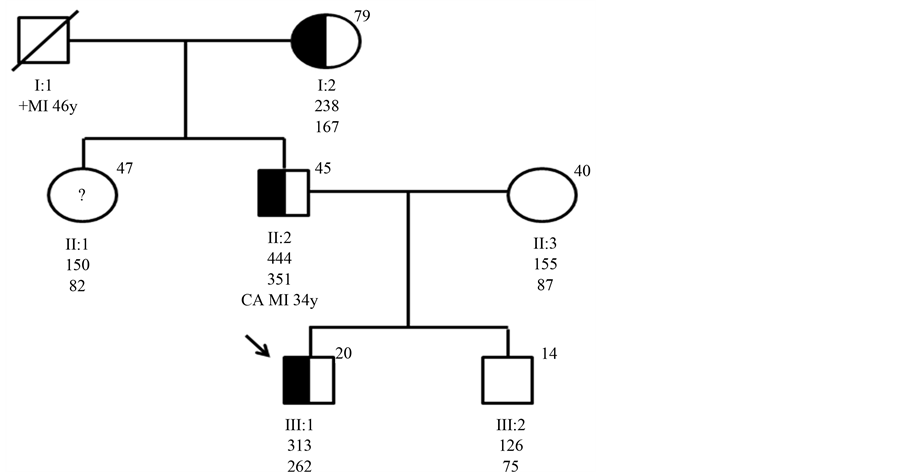
Figure 2. Genealogical tree of the family studied. The index-case is indicated by an arrow. Ages in years are shown in the upper right corner of the symbol. Total cholesterol and LDL-C values in mg/dL are shown below each symbol. Half shaped symbol correspond to family members with c.818-3C > G mutation. Open symbols represent individuals without the mutation. Open symbols with question mark represent individual not available for the study. Abbreviations, deceased; MI, myocardial infarction; CA, corneal arcus.
The genetic study showed the existence of a novel heterozygous point mutation—the Azorean-1. This mutation occurs in intron 5 of the LDLR gene (c.818-3C > G), causing a premature termination of transcription (stop codon). The family study was performed and it was confirmed that the mutation co-segregated in family members who are hypercholesterolemic (Figure 2).
3. Discussion
The study of familial hypercholesterolemia is extremely important because early changes of atherosclerosis have been demonstrated in the vasculature of children in the first two decades of life. On the other hand, from a socioeconomic point of view, early identification, diagnosis and appropriate treatment can significantly reduce the number of cardiovascular events and prevent premature death, which have a heavy impact on families and society.
In children, not always the suspected diagnosis is easy, particularly when the child is followed by another disease, which may also be responsible for hypercholesterolemia, as demonstrated in our case. Hypercholesterolemia is usually the only clinical finding in children with heterozygous FH in their first decade of life.
The serum cholesterol may be raised above 250 mg in hypopituitary children, even when there is no evidence of hypothyroidism, or even when hypothyroidism is adequately treated. Moreover, it was known that growth hormone induces low-density lipoprotein clearance, so its replacement in hypopituitarism reduces plasma LDLC by inducing LDL clearance [13] .
The genetic study of relatives of affected individuals is undoubtedly the most cost-effective method of detecting new cases of FH and several countries have already begun cascade screening programs [14] -[18] . Furthermore, clinical characterization is not sufficient to distinguish between patients with genetic or environmental dyslipidemia, and so molecular diagnosis is useful in clinical practice, allowing correct identification of FH patients and their relatives [17] [19] [20] . The grandmother of our index-patient (Figure 2) illustrated the importance of this molecular diagnosis, as she has total cholesterol below 290 mg/dL (90 - 190 mg/dL), easily controlled with statins, expressing a less aggressive phenotype. This molecular finding reveals that many individuals are probably being under diagnosed and have an even greater risk of developing premature CD.
As there is no record of grandfather cholesterol levels, we cannot state that he also suffered of FH, so the etiology of their MI might not be related to hypercholesterolemia.
The same mutation resulted in different phenotypes, as observed in the three family generations affected (Figure 2). In fact the index-patient father presents a more severe disease, with high cholesterol levels and difficult therapeutic control. This difference can be attributed to splicing mutation that produces a stop codon at the signal peptide, and consequently the protein is not translated. The amount of properly processed transcripts in patients with splice mutation is variable from person to person, giving a different intrafamilial variability in the clinical expression of FH. Another hypothesis is that the father being a compound heterozygote with another mutation, not screened in the study of the most frequent genes or never described. On the other hand, selected factors (such as the elevated apolipoprotein (a) levels, low levels of HDL-C and raised triglyceride and apolipoprotein B levels) were associated with a greater risk of developing CD [21] , as seen in index case father.
More than 1000 mutations have been described for the LDLR gene in a wide variety of populations [19] . In the last ten years, the Portuguese FH Study identified 80 different mutations in the LDLR gene, in 165 unrelated index patients, 34 being exclusive to the Portuguese population [22] . The mutation found in the LDLR gene of our family has not been previously described in the literature, and is to date exclusive to our study population, so we call it Azorean-1 mutation.
Since 1990’s, researchers began to examine the potential benefit of statin therapy in FH children on the premise that it prevent early onset atherosclerosis and reduce the increased risk of CD, when the effects of lifestyle modification are not sufficient.
As expected, statins are highly efficient in reducing plasma levels of LDL-C and have been proven to inhibit progression of early atherosclerotic changes in the vessel wall. But we still lack information about early and late-onset side effects of this class of drug, resulting from their early-onset and predicted life-long usage. The possible effects on mental development are related to the importance of cholesterol in the developing brain, for myelin formation, synaptogenesis, and neural plasticity. Other questions are the dosing used in FH children and the age at which statin therapy should be initiated [23] -[25] .
The Cochrane Review recommended that statin treatment should not be started before the age of eight, and that the duration of a delay in the initiation of statin therapy should be based on individual risk stratification [23] . To determine whether statins are safe as a long-term therapy in childhood, would require large and long-term randomized controlled multicenter trials with thousands of FH children, and of at least 10 years of follow-up [25] .
In children where statins are poorly tolerated or contraindicated, alternative lipid-lowering treatment might be necessary. Ezetimibe as a single drug might be an important treatment strategy for young patiens, although additional studies are needed to evaluate long-term safety and efficacy of Ezetimibe [25] [26] .
An important challenge is related to the education of physicians. A recent follow-up study showed that only 26% of children aged 10.9 (SD 4.2) years with FH were medicated with statins, while the remaining received lifestyle advice solely [27] . Since FH inevitably leads to premature CD, only full compliance with statins is an adequate goal [24] . Pediatricians and general physicians should be aware of the benefits of the early identification of these patients and make use of molecular diagnosis to correctly treat their cardiovascular risk.
4. Conclusions
It was very important to establish a Portugal-wide register of children with a clinical and genetic diagnosis of FH with annual follow-up, in order to learn about the new mutations and their impact in the different phenotypes and therapeutic efficiencies.
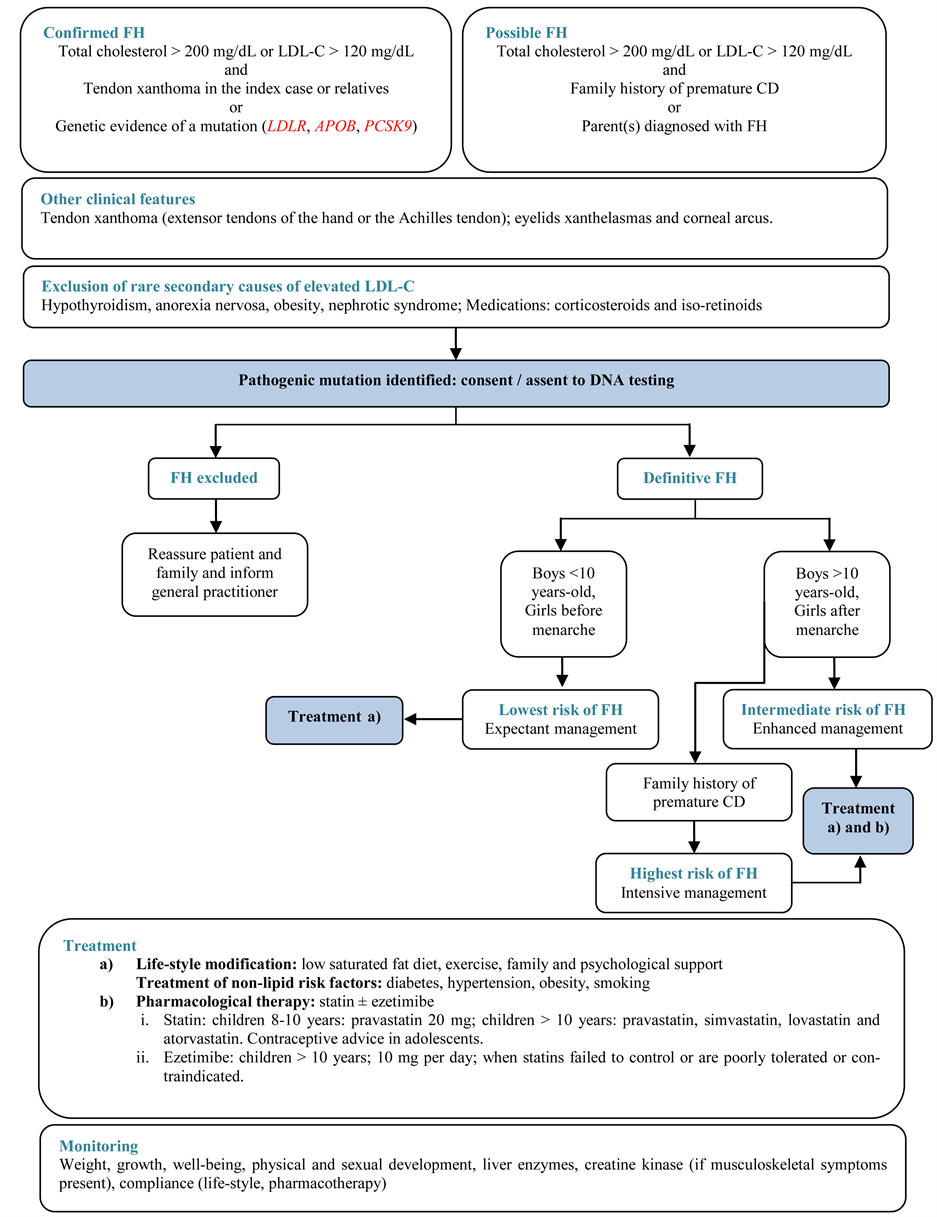
Finally, childhood is an important time to optimize cardiovascular health because risk factors manifest when health behaviors develop and risk reduction will have the greatest impact. All individuals with a possible diagnosis of FH should be clinically assessed according to the recommended options shown in the algorithm.
Supplementary Figure. Algorithm for the Diagnosis and Management of FH in Children and Adolescents.
NOTES
*Corresponding author.