1. INTRODUCTION
Nicotine, the main substance responsible for tobacco dependence [1], interacts with nicotinic acetylcholine receptors (nAChRs) and modulates functions such as attention, learning, and memory; furthermore, nAChRs have also been implicated in certain pathologies, including Parkinson’s and Alzheimer´s diseases [2,3]. Besides activating nAChRs, nicotine also inhibits K+ channels. This last effect has been studied principally in myocytes and heterologous expression systems [4-9].
In the brain, nicotine acts on neurons and astrocytes [10-12], enhancing hippocampal synaptic transmission and long-term memory [13,14]. Furthermore, activation of astrocytic nAChRs increases their intracellular Ca2+ concentration ([Ca2+]i) [12,15-17]; upregulating the expression of the glial derived neurotrophic factor implicated in neuroprotection [18], and increasing S100B secretion, a possible indicator of brain injury [19]. It is known that glial Ca2+ signaling is triggered by activation of multiple metabotropic and ionotropic transmitter receptors, and this signal is an important way for neurons and astrocytes to communicate [16,20-24].
Previously, we found that nicotine activates nAChRs and inhibits non-decaying K+ currents on hippocampal astrocytes [10]. Therefore, in present study we explored whether these two different actions of nicotine, as nAChR agonist and K+ channel inhibitor, result in changes in the [Ca2+]i in astrocytes that would modulate their function in the central nervous system.
2. METHODS
All procedures were carried out in strict accordance with the recommendations of the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the local Animal Research Committee of the Instituto de Neurobiología at Universidad Nacional Autónoma de México.
2.1. Cell Cultures
Astrocyte cultures were obtained as previously described [10]. Two newborn Wistar rats were decapitated and pithed. The brains were removed and placed in cold recording solution (in mM): 136 NaCl, 2.5 KCl, 2 CaCl2, 0.5 MgCl2, 10 HEPES, and 10 D-glucose, pH 7.4. Then, under stereomicroscope, coronal brains slices were obtained and the hippocampus was dissected. Next, tissue was cut into small pieces and then mechanically dissociated until obtain isolated cells. The cell suspension was placed in a 35-mm Petri dish on a glass coverslip coated with 0.01% poly-L-ornithine. Then, we added 2 mL Dulbecco’s Modified Eagles Medium supplemented with 10% fetal bovine serum, 2 mM glutamine, 100 units/mL penicillin, and 100 μg/mL streptomycin. For astrocyte selection after 24 h of culture, the medium was replaced by serum-free neurobasal medium with 2 mM glutamine, 100 units/mL penicillin, 100 μg/mL streptomycin, and G5 supplement. The medium was changed every four days. All culture reagents were purchased from Gibco (Carlsbad, CA, USA).
2.2. Fluorescence Measurements
To record [Ca2+]i increases, astrocytes (cultured for 4 - 7 days) were loaded with 10 mM fluo-4 acetoxymethyl ester (fluo-4 AM) for 30 - 40 min at room temperature, and then washed for 10 min with the recording solution. Cells were placed in a chamber (volume ~500 mL) continuously perfused (4 mL/min) with the recording solution, and were recorded with a LSM 510 (Carl Zeiss) laser confocal system. Fluo-4 AM was excited with the 488 nm line of an argon laser, and the emitted fluorescence was collected at wavelengths from 505 to 550 nm through an objective with 0.8 of numerical aperture. Images were acquired at 1 Hz. Changes in fluorescence (ΔF) were measured as the relative increase from baseline fluorescence in the whole recorded astrocyte and expressed as %ΔF/F0 = 100[(Fpost − Frest)/Frest]. Ca2+ increases were measured at the maximal level reached during each recording. Astrocytes were considered as responsive when, after stimulus, fluorescence signal increased at least three times the standard error of the average basal fluorescence. The results are shown as %ΔF/F0. Comparison of the mean values was performed by the t-Student or one way analysis of variance (ANOVA) followed by Tukey’s post-test; p < 0.05 was considered statistically significant.
Local application of drugs for 2 s was carried out with a U-tube system, commanded by a solenoid valve, placed at ~250 µm from the recording cell. In experiments with long exposure time the substances were added to the recording solution. All the drugs were prepared as concentrated stocks in distilled water and stored frozen. The day of use, drugs were diluted in the recording solution.
3. RESULTS
3.1. nAChR Agonists Induced Intracellular Ca2+ Increments
Confluent cell cultures (n = 5) were incubated with an antibody against glial fibrillary acidic protein, and more than 99% of the cells were immunopositive, indicating their astrocytic lineage (data not shown; see [10]). Astrocytes display intra and intercellular spontaneous Ca2+ signaling [24]. However, to study direct actions of nicotine on astrocytic membrane channels that in turn change the [Ca2+]i, we selected completely isolated astrocytes without apparent Ca2+ spontaneous activity previous to stimulation with drugs. Thus, in 13 out 37 astrocytes (n = 4 cultures) the application of 10 mM nicotine resulted in an increase of the fluo-4 fluorescence by 68 ± 18%. Moreover, 99 out of 190 astrocytes (n = 32 cultures) responded to 100 mM nicotine, increasing by 82 ± 11% ΔF/F0. Finally, a high concentration of nicotine (1 mM) elicited in eight out of 11 astrocytes (n = 7 cultures) a fluorescence increase by 85 ± 5% (Figures 1(A)-(E)). The three concentrations of nicotine lead to similar mean amplitude responses with no statistically significant differences (Figure 1(E)). Next, for most experiments 100 mM nicotine was used. Astrocytes responded to nicotine with two different profiles. About the 50% of responsive cells showed an initial rapid and transient elevation of [Ca2+]i that decayed to baseline after 5 min (Figures 1(A) and (C)). In the rest of cells, the [Ca2+]i increased gradually, without returning to the baseline by the end of the recording (Figures 1(B) and (D)). The two types of responses were obtained with the three nicotine concentrations tested (Figure 1(F)). Astrocytic [Ca2+]i increases elicited by nicotine were totally prevented when Ca2+ was removed from the extracellular solution (Figure 1(E); n = 9, 3 cultures). In addition, no correlation was observed between the number of days (4 - 7) the astrocytes had been in culture and the type of Ca2+ response evoked by nicotine.
It is known that hippocampal astrocytes express nAChRs [10,16,17], which are permeable to Ca2+ [26]. Therefore, the increase of [Ca2+]i in response to nicotine is likely due to the activation of nAChRs. In this sense, we applied two other nAChRs agonists, acetylcholine and choline (Figure 1(G)). Fifteen out of 19 astrocytes responded to the application of 100 mM acetylcholine (in the presence of 500 nM atropine to block muscarinic receptors) with an increase in [Ca2+]i of 38 ± 5% (n = 3

Figure 1. Nicotine-induced intracellular Ca2+ increases in hippocampal astrocytes. ((A), (B)) Representative fluorescence images obtained from two astrocytes, before (0 s) and at maximal fluorescence signal after application of 100 mM nicotine. ((C), (D)) Corresponding examples and average nicotine-induced rapid transients (n = 50), or slow and gradually Ca2+ increases (n = 49) recorded from astrocytes as those illustrated in A and B, respectively. (E) Summary of data as the mean ± S.E.M. of the %ΔF/F0 elicited by nicotine (NIC); including those without extracellular Ca2+ ([Ca2+]o = 0, ANOVA and Tukey post-test; *p < 0.05). (F) Percentage of astrocytes responding to different concentrations of nicotine. Grey and white areas correspond to the percentage of cells with [Ca2+]i signal profile similar to (C) and (D), respectively. (G) Images and corresponding Ca2+ responses evoked by nicotine, choline (Ch), and acetylcholine (ACh) recorded in the same astrocyte. Drugs were washed out for 5 min. (D) Summary of data of the %ΔF/F0 elicited by nAChR agonists. Responses elicited by nicotine were significantly higher than those for acetylcholine (*) and choline (#) (t-Student test, p < 0.05). (E) Percentage of astrocytes that responded to nAChR agonists. Scale bar: 10 mm; arrows indicate the 2-s drug application.
cultures), whereas 14 out of 19 astrocytes responded to 100 mM choline with a 31 ± 6% increase of [Ca2+]i (n = 3 cultures). Nicotine (at 100 µM) produced significantly higher [Ca2+]i increases with respect to those elicited by the other nAChR agonists at the same concentration. However, the percentage of astrocytes that responded to acetylcholine and choline was greater compared with those for nicotine (Figure 1(H)). The delay between the application of substances and the onset of the Ca2+ increment was highly variable, and no significant differences were found throughout the experiments: 37 ± 12 s for nicotine (n = 99), 33 ± 15 s for acetylcholine (n = 15), and 23 ± 17 s for choline (n = 14), all at 100 μM.
3.2. Nicotine Increases Intracellular Ca2+ Levels Independently on nAChRs
In addition to the activation of nAChRs, nicotine also inhibits K+ currents, leading to membrane depolarization [4-11], which might open voltage-gated Ca2+ channels that favor the influx of Ca2+ ions [26]. Thus, to evaluate this possibility, astrocytes were superfused for at least 10 min with a cocktail of nAChRs antagonists (100 mM mecamylamine, 10 nM methyllycaconitine, and 10 nM dihydro-b-erythroidine) to eliminate nAChRs activation. After the incubation with antagonist, cells were challenged with nicotine (100 mM; 5 s). In control conditions, i.e., without nAChR antagonists, nicotine increased [Ca2+]i by 88 ± 10% in 17 out of 49 astrocytes. These responses were similar to those previously observed (Figure 1). Interestingly, when the cocktail of antagonists for nAChRs was present, in 10 out of 38 astrocytes, nicotine generated [Ca2+]i increases of 31 ± 3% (Figures 2(A) and (B); n = 4 cultures). Furthermore, in some astrocytes (n = 7) nicotine generated Ca2+ responses only in the absence of nAChR antagonists (Figure 2(B)), indicating that they were due to the activation of nAChRs. Finally, in other astrocytes (n = 7), nicotine did not generate any Ca2+ responses, neither in the presence nor in the absence of nAChR antagonists (Figure 2(C)). In those astrocytes that responded to nicotine in the absence and the presence of nAChR antagonists, the percentage of responsive astrocytes was similar in both experimental conditions (34.7 and 26.3%, respectively), but the mean amplitude of Ca2+ responses were significantly different (see above, 88 vs. 31% DF/F0, respectively).
Nicotine inhibits non-decaying K+ currents in astrocytes [10], effect that might result in membrane depolarization that activates voltage-gated Ca2+ channels [26], and increments of the [Ca2+]i. Thus, we explored the effect of the inhibition of K+ currents with tetraethylammonium (broad spectrum blocker of K+ channels [27]) on astrocytic [Ca2+]i. In 10 out of 33 astrocytes (n = 3 cultures), the application of 20 mM tetraethylammonium increased the fluorescence by 86 ± 15%. Then, these astrocytes were superfused with nAChR antagonists and nicotine was applied, resulting again in increases of [Ca2+]i (Figure 2(D)). Additionally, astrocyte membrane was depolarized by application of a high concentration of extracellular K+ (75 mM, 5 s), and in four out of nine astrocytes the fluorescence increased by 51 ± 12% (data not shown). Together, these results indicate that the inhibition of K+ currents in astrocytes, either by nicotine or tetraethylammonium, as well as membrane depolari-
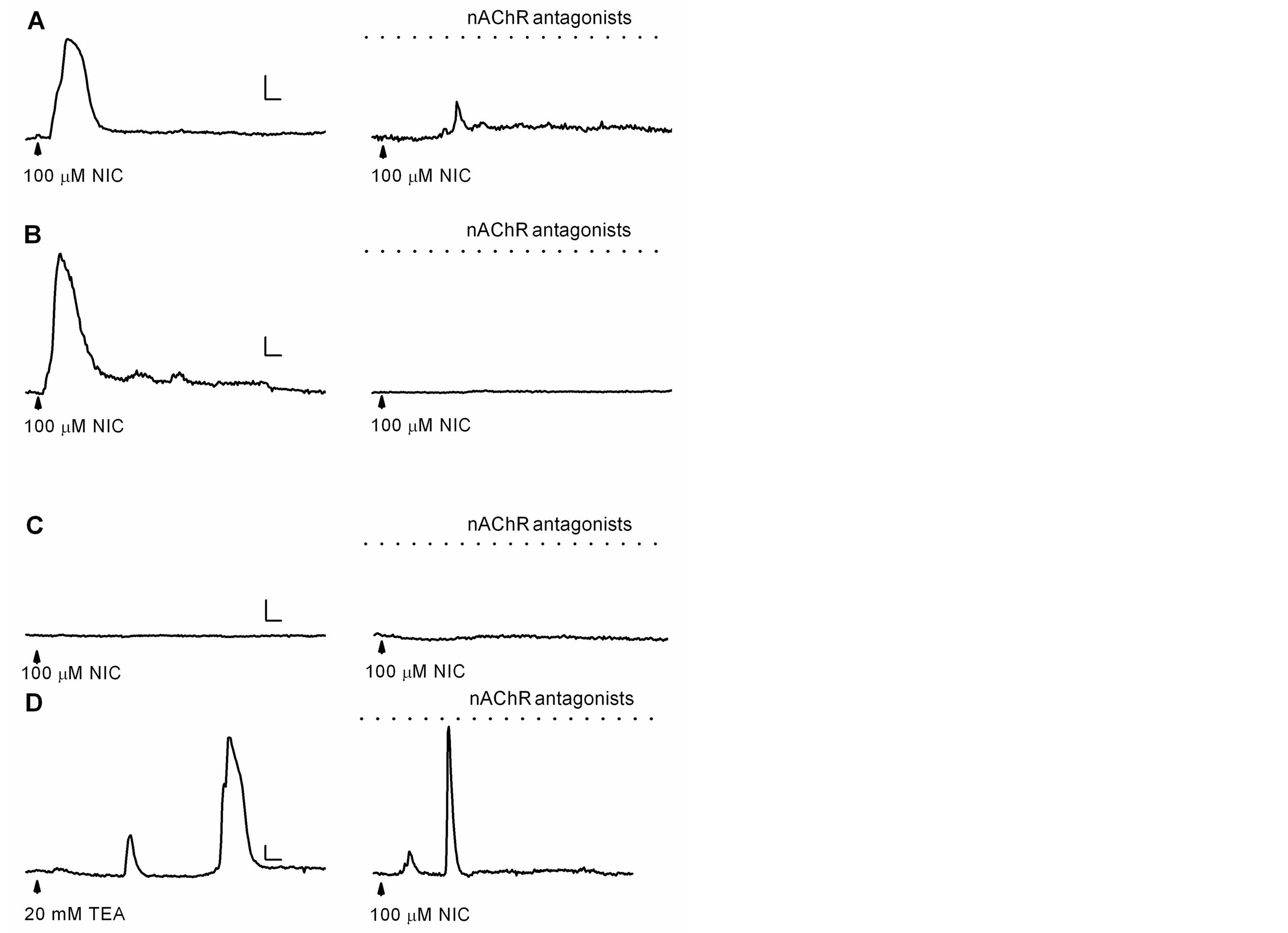
Figure 2. Ca2+ responses generated by nicotine or tetraethyllammonium. ((A)-(C)) Ca2+ responses generated by nicotine in the absence (left) or presence (right) of the nAChR antagonist cocktail (dotted line: 100 mM mecamylamine, 10 nM methyllycaconitine, and 10 nM dihydro-b-erythroidine) in three different astrocytes; (D) Ca2+ responses generated by tetraethylammonium (left) and nicotine in the presence of the nAChR antagonist cocktail (right). Horizontal bars: 25 s, vertical bars: 20% ΔF/F0 (A), 40% ΔF/F0 ((B)-(D)).
zation by high extracellular K+, all resulted in increases of the [Ca2+]i.
Nicotine applied at a low concentration (100 nM) and over a relatively long time (15 min) inhibited K+ currents in hippocampal astrocytes by ~43% [10]. Using these same conditions, we evaluated changes in [Ca2+]i levels. In 19 out of 32 astrocytes tested, nicotine increased [Ca2+]i by 92 ± 11%. Some astrocytes (n = 5) show rapid and transient [Ca2+]i increases, whereas in other astrocytes (n = 9), nicotine generated gradual elevations of [Ca2+]i. Additionally, in the rest of astrocytes (n = 5), nicotine generated responses with more than two rapid and transient [Ca2+]i increases (Figure 3).
4. DISCUSSION
In the present work we showed that astrocytic Ca2+ responses generated by nicotine were consequence of: a) nAChRs activation, b) K+ currents inhibition, because
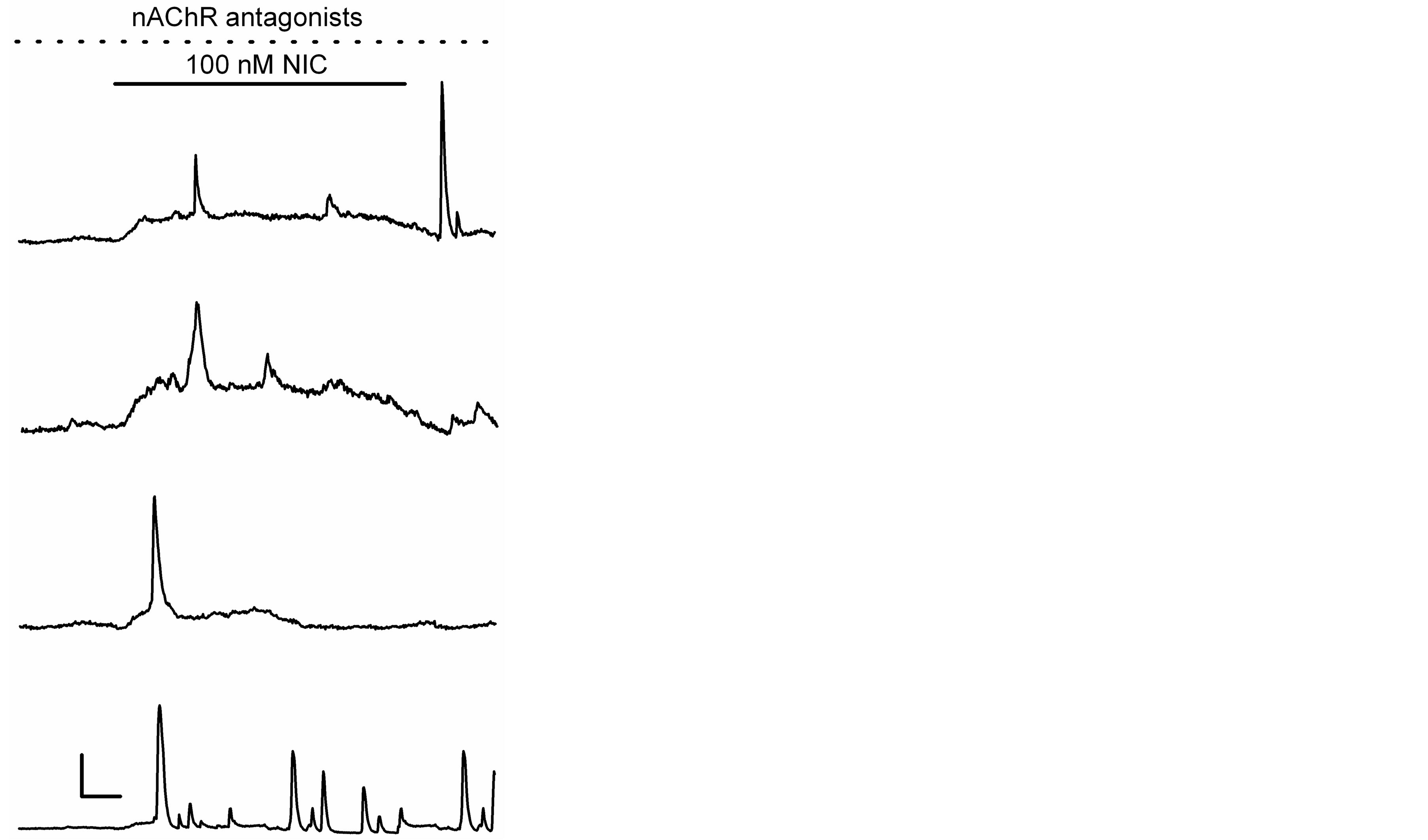
Figure 3. Ca2+ responses generated by a low concentration of nicotine. Ca2+ responses induced by nicotine during 15 min (solid line) in the continuous presence of nAChR antagonists (as in Figure 2), in four different astrocytes. Horizontal bar: 3 min, vertical bar: 30% ΔF/F0 (two upper records) and 40% ΔF/F0 (two bottom records).
nicotine in the presence of antagonists for different types of nAChRs (mecamylamine, methyllycaconitine plus dihydro-b-erythroidine; [28]) increased the [Ca2+]i, and c) a combination of both effects in the same cell, because in some astrocytes the amplitude of nicotine-induced Ca2+ responses in the presence of nAChR antagonists were lower than those in the absence of antagonists (see Figure 2(A)). To highlight is the finding that nicotine affect [Ca2+]i in astrocytes by a different pathway from its typical action as nAChR agonist.
We think that nicotine-induced [Ca2+]i signals are strongly related with nicotine-induced electrical responses described in a previous study, namely the activation of nAChRs and the inhibition of a non-decaying K+ current, [10]. In the former case, the [Ca2+]i may increase by the influx of Ca2+ through the ion channels of nAChRs, and in turn amplified by triggering Ca2+-induced Ca2+-release from intracellular stores, store-operated Ca2+ entry, or the opening of voltage-gated Ca2+ channels [16,24,25,29]. In the latter case, inhibition of K+ channels could depolarize the astrocytes, and consequently the activation of voltage-gated Ca2+ channels, expressed in astrocytes [26,30,31], allowing Ca2+ to enter the cell. In support of the latter, tetraethylammonium also increased the astrocytic [Ca2+]i (Figure 2(D)).
The present work is consistent with the evidence that glial cells express nAChRs [10-12,16-18,32-36]. These glial receptors are activated by nAChRs agonists, resulting in increases of the [Ca2+]i [16,17], and it is very likely that these receptors are involved in glial cell functions, including bidirectional glia-neuron communication. For instance, neuronal and glial nAChRs participate in modulating both hippocampal synaptic transmission and long-term memory [14,37,38], neuroprotection, and inflammation [18,19,39].
The astrocytic nAChRs and K+ channels may be reached by nicotine during smoking [1,40]. Here we found that long exposure (15 min) to 100 nM nicotine in the presence of nAChR antagonists increased the [Ca2+]i, likely by inhibiting K+ currents [10]. This finding coincides with reports that have shown that nicotine is a non-specific inhibitor of K+ channels, and exerts their action even at very low concentrations as in the nanomolar range [4-9]. These results are relevant, considering that in regular smokers nicotine concentration in the venous blood is 60 - 300 nM, whereas in the arterial blood, which better represents the nicotine level in the brain, it is 600 nM, with a half-life close to 2 h [1,40].
Thus, nicotine seems to have more sites of action in the brain than previously thought [10,41]. In accordance, the present work shows that nicotine partly exerts its effect in the astrocytic [Ca2+]i in a nAChR-independent mode. The unmasked inhibitory effect of this drug on K+ channels, in addition to their well known action as nAChR agonist, may account for the wide variety of effects of nicotine on both neuronal and glial cells. However, further studies in brain slices are needed to corroborate the actions of nicotine on astrocytic Ca2+ activity, and their impact on brain functions.
The involvement of nAChRs, as well as nicotine actions, have become increasingly relevant in certain physiological and pathological conditions such as memory and cognition, epilepsy, schizophrenia, and Alzheimer’s disease [2,3], in which astrocytes and their release of gliotransmitters, such as D-serine, have been implicated [14,42,43].
5. CONCLUSION
The present study shows that nicotine, acetylcholine, and choline, as well as tetraethylammonium, and depolarization by high extracellular K+ concentration, result in increases of the [Ca2+]i in hippocampal astrocytes. The actions of nicotine are due to activation of nAChRs and/or inhibition of K+ channels. Thus, through affecting two different target membrane channels, nicotine impacts the astrocytic functions and probably play their role in cholinergic synaptic transmission and also in processes as synaptic plasticity, learning, memory, nicotine addiction, among others.
ACKNOWLEDGEMENTS
This work was supported by a Grant from Dirección General de Asuntos del Personal Académico IN201313 to J.G.C. We are grateful to Martín García Servín for help in caring for the rats and to Dra. Marcela Miranda-Morales for technical assistance. We express our gratitude to Dra. Dorothy Pless for critically reviewing the manuscript.
NOTES