Docking Studies, Synthesis, and Evaluation of Antioxidant Activities of N-Alkylated, 1,2,4-Triazole, 1,3,4-Oxa-, and Thiadiazole Containing the Aminopyrazolopyridine Derivatives ()
1. Introduction
1H-Pyrazolo[3,4-b]pyridines comprise a very interesting class of compounds because of their significant and versatile biological and pharmacological activities, such as antimalarial [1] antiproliferative [2] antimicrobial [3-5] inhibition of cyclin-dependent kinases [6] and cardiovascular [7-9] antiviral [10-12] and antileishmanial [13] activities. Pyrazole fused pyridines and pyrimidines are known to possess a wide range of biological activity. Specifically pyrazolopyridines exhibit antitubercular, anxiolytic [14].
It has been reported that certain compounds bearing 1,3,4-oxa-, thiadiazole, and 1,2,4-triazole nucleus possess significant anti-inflammatory activity [15,16]. Also, in view of these reports and in continuation of our recent work on the pyrazolo[3,4-b]pyridine derivatives, to synthesize a new heterocyclic compounds [17,18], we reported here the synthesis of a number of new alkylated 4,6-dimethyl-1H-pyrazolo[3,4-b]pyridine-3-amine, formation of 1,2,4-triazoles, 1,3,4-oxadiazole, and 1,3,4-thiadiazoles.
Design of drug targets containing two carboxylic groups appropriately attached to the opposite sides of the aromatic fragment (e.g., naphthalene ring) as shown by our synthesized compounds to interact with the active site hydrophobic pocket while the first carboxylate may occupy the oxyanion hole and the second forms H-bonds with oxygen of coenzyme’s diphosphate moiety will be a good rational for potent inhibitors.
2. Material and Methods
2.1. Experimental
Melting points were determined on a Buchi melting point. IR spectra were recorded with a Perkin-Elmer model 1720 FTIR (KBr), 1H NMR spectra were recorded with Bruker AC 250 FT NMR spectrometer at 250 MHz with TMS as an internal standard. EIMS and FABMS spectra were recorded with a Finnigen MAT 312 = AMD. The microanalyses were performed at the microanalytical unit, Cairo University.
Ethyl 2-(3-amino-4,6-dimethyl-1H-pyrazolo[3,4-b] pyridin-1-yl)acetate (2). To a stirred suspension of pyrazolopyridine 1 (0.81 g, 5 mmol) in dry DMF (10 mL) contaning K2CO3 (0.67 g, 5 mmol), the ethyl chloroacetae (0.61 g, 5 mmol) was added dropwise. The reaction mixture was stirred at room temperature for an additional 13 h and then poured into ice cold water with stirring. The obtained solid product was collected by the filtration, washed with water and recrystallized from ethanol to afford the pale yellow crystals of ester 2. (0.77 g, 95%), m.p. 127˚C - 129˚C. IR (KBr, νmax, cm−1): 3316 (NH2), 2923, 2853 (CH aliphatic), 1722 (C=O), 1596 (C=N). 1H NMR (CDCl3), δ, ppm: 1.13 (3H, t, J = 7 Hz, CH3CH2), 4.22 (2H, q, J = 13 Hz, CH3CH2), 2.70 (3H, s, CH3), 2.79 (3H, s, CH3), 5.09 (2H, s, CH2), 6.57 (1H, s, H-5), 7.14 (2H, bs, NH2). Mass spectrum, m/z (I, %): 249 [M++1] (35), 248 [M+] (100), 161 (70), 131 (16). Found, %: C 58.05; H 6.50; N 22.57. C12H16N4O2 Calculated, %: C 57.99; H 6.22; N 22.32.
2-(3-Amino-4,6-dimethyl-1H-pyrazolo[3,4-b] pyridin-1-yl) acetohydrazide (3). To a suspension of ester 2 (1.24 g, 5 mmol) in ethanol (15 ml), an excess of hydrazine hydrate (4 ml) was added. The reaction mixture was refluxed for 4 h , cooled, the solid product was collected by filteration, deried, and recrystalized from methanol to give the pale yellow crystals of compound 3 (0.91 g, 77%). mp 150˚C - 152˚C IR (KBr) (cm−1), ν = 3343 - 3202 (NHNH2), 2938, 2892 (CH aliphatic), 1660 (C=O), 1589 (C=N). 1H NMR (DMSO-d6), δ, ppm: 2.45 (3H, s, CH3), 2.51 (3H, s, CH3), 4.98 (2H, s, CH2), 5.12 (2H, bs, NH2NH), 6.54 (1H, s, CH-5), 6.79 (2H, bs, NH2), 9.16 (1H, bs, NH2NH). Mass spectrum, m/z (I, %): 234 [M+] (8), 252 (25), 219 (23), 175 (100). Found, %: C 51.27; H 6.02; N 35.88. C10H14N6O Calculated, %: C 51.13; H 5.88; N 35.47.
5-((3-amino-4,6-dimethyl-1H-pyrazolo[3,4-b] pyridin-1-yl)methyl)-1,3,4-oxadiazole-2-thiol (4). Hydrazid 3 (0.94 g, 4 mmol) and CS2 (0.3 g, 4 mmol) were added to a solution of KOH (0.22 g, 4 mmol) in water (10 ml) and ethanol (10 ml). The reaction mixture was refluxed for 4 h. After evaporation under reduced pressure, a solid was obtained. This was dissolved in water and acidified with conc. HCl. The precipitate was filtered, washed with water, and recrystallized from methanol to give colorless powder, yield 0.72 g, 65%, mp 230˚C - 233˚C (MeOH). IR spectrum (thin layer), ν, cm−1: 2784, 2725 (S˗H), 1566 (C=N). 1H NMR spectrum (300 MHz, DMSO-d6), δ, ppm (J, Hz): 2.45 (3H, s, CH3), 2.52 (3H, s, CH3), 4.18 (s, 2H, CH2), 6.74 (1H, s, CH-5), 6.87 (2H, bs, NH2) 13.82 (brs, 1H, SH). 13C NMR spectrum (75.5 MHz, DMSO-d6), δ, ppm: 15.13, 19.62 (2 CH3), 33.30 (CH2), 115.12, 125.17, 128.59, 128.79, 129.37, 129.72, 135.84, 144.28 (Ar-C). Mass spectrum (EI, 70 ev), m/z (Irel, %): 277 [M++ 1] (2), 276 [M+] (20), 194 (18), 161 (24), 129 (22), 44 (74), 28 (100). Found, %: C, 47.81, H, 4.38, N, 30.41. C11H12N6OS.Calculated, %: C, 47.32; H, 4.31; N, 30.29.
4,6-dimethyl-1-((5-(alkylthio)-1,3,4-oxadiazol-2-yl)methyl)-1H-pyrazolo[3,4-b]pyridin-3-amine (5a-c). To a stirred suspension of oxadiazol thion 4 (1mmol) in dry DMF (10 ml), sodium hydride (0.04 g, 1 mmol, 60% dispersion oil) was added. When liberation of hydrogen had ceased (1.5 h), the appropriate alkyl reagent (ethyl-, methyl iodide and benzylchloride) was added in dropwise, and the reaction mixture was stirred at room temperature for 10 - 15 h. The solvent was removed under reduced pressure and the residue was triturated with cold water with stirring for 2 - 5 h, the products was filtered off, deride and recrystallized with ethanol to give 5a-c.
1-((5-(Ethylthio)-1,3,4-oxadiazol-2-yl)methyl)-4,6-dimethyl-1H-pyrazolo[3,4-b]pyridin-3-amine (5a). Colorless crystals, yield 0.19 g 61%, mp 139˚C -140˚C; 1H NMR spectrum (300 MHz, CDCl3), δ, ppm (J, Hz): 0.93 (3H, t, J = 7.0 Hz, CH3CH2), 2.45 (3H, s, CH3), 2.53 (3H, s, CH3), 3.11 (2H, q, J = 7.1, SCH2CH3), 4.87 (2H, s, N-CH2), 6.88 (1H, s, H-5), 7.02 (2H, bs, NH2). Mass spectrum (EI, 70 ev), m/z (Irel, %): 304 [M+] (20), 290 [M-CH3], 148 (100). Found, %: C, 51.30; H, 5.30; N, 27.16. C13H16N6OS. Calculated, %: C, 51.15; H, 5.14; N, 27.09.
4,6-Dimethyl-1-((5-(methylthio)-1,3,4-oxadiazol-2-yl)methyl)-1H-pyrazolo[3,4-b]pyridin-3-amine (5b). Colorless crystals, yield 0.24 g, 77%, mp 160˚C - 162˚C; 1H NMR spectrum (300 MHz, CDCl3), δ, ppm (J, Hz): 2.33 (3H, s, CH3), 2.49 (3H, s, SCH3), 2.54 (3H, s, CH3) , 4.55 (2H, s, N-CH2), 6.67 (1H, s, H-5), 7.25 (2H, bs, NH2). Mass spectrum (EI, 70 ev), m/z (Irel, %): 290 (M+, 43), 148 (100). Found, %: C, 49.64; H, 4.86; N, 28.95. C12H14N6OS. Calculated, %: C, 49.45; H, 4.52; N, 28.65.
1-((5-(benzylthio)-1,3,4-oxadiazol-2-yl)methyl)-4,6-dimethyl-1H-pyrazolo[3,4-b]pyridin-3-amine (5c). Colorless powder, yield 0.31 g, 78%, mp 202˚C - 204˚C; H1 NMR spectrum (300 MHz, CDCl3), δ, ppm (J, Hz): 2.53, 2.64 (6H, 2s, 2CH3), 4.40 (2H, s, SCH2Ph), 4.55 (2H, s, CH2), 6.32 (1H, s, H-5), 6.67 - 7.47 (5H, m, Ph). Mass spectrum (EI, 70 ev), m/z (Irel, %): 366 [M+], 289 (55), 275 (34), 91 (100). Found, %: C, 59.00; H, 4.95; N, 22.93. C18H18N6OS. Caculated, %: C, 58.87; H, 4.34; N, 22.86.
2-(2-(3-Amino-4,6-dimethyl-1H-pyrazolo[3,4-b] pyridin-1-yl)acetyl)-N-phenylhydrazinecarbothioamide (6). To suspension of acid hydrazide 3 (2.34 g, 10 mmol) in absolute ethanol (20 mL), PhNCS (1.35 g, 10 mmol) was added. The reaction mixture was heated under reflux for 6 h. The product that separated on cooling was filtered off, washed with ethanol dried and recrystallized from methanol to give a colorless crystals of compound 6, yield, 3.46 g, 94%, m.p. 173˚C - 175˚C. IR (KBr, νmax, cm−1): 3336 - 3244 (NH, NH2), 4035, 2978, 2886 (CH aliphatic), 1697 (CONH), 1593 (C=N), 1195 - 1130 (C=S). 1H NMR (CDCl3), δ, ppm: 2.55 (3H, s, CH3), 2.68 (3H, s, CH3), 4.95 (2H, s, CH2), 6.98 (1H, s, H-5), 7.93-8.54 (7H, m, NH, ph), 8.77 (1H, s, N=CH), 9.07 (1H, bs, CSNH), 10.43 (1H, bs, CONH). Mass spectrum, m/z (I, %): 270 [M++1, 20], 269 [M+, 70], 192 (35), 240 [M+] (100), 161 (70), 131 (16). Found, %: C 55.27; H 5.18; N 26.54. C17H19N7OS Calculated, %: C 55.33; H 5.15; N 26.57.
5-((3-amino-2-yl-methyleneamino)-4,6-dimethyl-1H-pyrazolo[3,4-b]pyridin-1-yl)methyl)-N-phenyl-1,3,4-thiadiazol-2-amine (7). The phenylhydrazinecarbothioamide 6 (1.34 g, 5 mmol) was added gradually with stirring to cold conc. H2SO4 (10 ml) during 10 min. The mixture was further stirred for another 2 h in an ice bath. The solid separated out was filtered, washed with water, dried and recrystallized from methanol to give a yellow crystals of compound 7, yield, (2.95 g, 84%), m.p. 190˚C -192˚C. IR (KBr, νmax, cm−1): 3316 - 3285 (NH), 2955 - 2933 (CH aliphatic), 1598 (C=N). 1H NMR (CDCl3), δ, ppm: 2.51 (3H, s, CH3), 2.64 (3H, s, CH3), 5.33 (2H, s, CH2), 6.33 (1H, s, H-5),7.21 (2H, bs, NH2) 7.55 - 7.76 (6H, m, ph), 11.55 (1H, bs, NHPh). Mass spectrum, m/z (I, %): 352 [M++1] (35), 351 [M+] (100), 161 (70), 131 (16). Found, %: C 58.01; H 4.88; N 27.90. C17H17N7S Calculated, %: C 58.17; H 4.74; N 27.72.
5-((3-amino-2-yl-methyleneamino)-4,6-dimethyl-1H-pyrazolo[3,4-b]pyridin-1-yl)methyl)-4-phenyl-4H-1,2,4-triazole-3-thiol (8). A suspension of thiosemicarbazide 8 (2.68 g, 10 mmol) in ethanol (50 ml) was dissolved in 4N aqueous sodium hydroxide (50 ml), resulting in the formation a clear solution. The reaction mixture was refluxed for 4 h on water bath, concentration, cooled, and filtered. The pH of the filtrate was adjusted between 5-6 with acetic acid and kept aside for 2 h. The solid separated out was filtered, washed with water, dried and recrystallized with ethanol to give white powder of compound 8, (2.35 g, 67%), m.p. 217˚C - 219˚C. IR (KBr, νmax, cm−1): (NH2), 3022 (CH aromatic), 2955-2933 (CH aliphatic), 2786 - 2715 (SH), 1638 (C=N). 1H NMR (CDCl3), δ, ppm: 2.50 (3H, s, CH3), 2.61 (3H, s, CH3), 5.39 (2H, s, CH2), 6.23 (1H, s, H-5), 7.22 - 7.75 (8H, m, ph, NH2), 12.05 (1H, bs, SH). Mass spectrum, m/z (I, %): 352 [M++1] (35), 351 [M+] (69), 160 (35), 131 (100). Found, %: C 58.01; H 4.88; N 27.90. C17H17N7S Calculated, %: C 58.28; H 4.95; N 27.66.
5-((3-amino-4,6-dimethyl-1H-pyrazolo[3,4-b]pyridin-1-yl)methyl)-N-phenyl-1,3,4-oxadiazol-2-amine (9): Mercuric oxide (2.37 g , 11 mmol) was added to solution of thiosemicarbazide 6 (2.69 g, 10 mmol) in methanol (20 mL) and the resulting mixture was refluxed for 3 h. The precipitate mercuric sulfide was filtered off and washed with hot methanol. The filtrate on cooling gave a solid product which was filtered, dried and recrystallized from methanol to give a yellow crystals of oxadiazol 9 (2.78 g, 83%), m.p. 210C˚ - 212˚C. IR (KBr, νmax, cm−1): 3230 - 3205 (NH, NH2), 3055 (CH aromatic), 2944 - 2910 (CH aliphatic), 1635 (C=N). 1H NMR (CDCl3), δ, ppm: 2.29 (3H, s, CH3), 2.50 (3H, s, CH3), 5.28 (2H, s, CH2), 6.13 (1H, s, H-5), 7.05 - 7.20 (8H, m, ph, NH2), 10.12 (1H, bs, NHPh). Mass spectrum, m/z (I, %): 336 [M++1] (35), 335 [M+] (100), 147 (70), 128 (16). Found, %: C 60.96; H 5.25; N 29.32. C17H17N7O Calculated, %: C 60.88; H 5.11; N 29.24.
2.2. Molecular Modeling Studies
2.2.1. Generation of Ligand and Enzyme Structures
The crystal structure of AKR1C3 complexed with its bound inhibitor Indomethacin was downloaded through the Protein Data Bank PDB/ RCSB site and saved as *.pdb file [18].
A set of novel N-Alkylated, 1,2,4-triazole, 1,3,4-oxa-, thiadiazole and complexes containing the aminopyrazolopyridine derivatives were designed to inhibit AKR1C3. All compounds were built in ChemDraw Ultra version 8.0.3 and their energy minimized through Chem3D Ultra version 8.0.3/ MM2, Jop Type: minimum RMS Gradient of 0.100, and saved as MDL MolFile (*.mol).
2.2.2. Docking Using Molsoft ICM 3.4-8C Program
The novel energy-minimized Indomethacin analogues were docked into the active site of AKR1C3 crystal structure using ICM-Pro software version 3.4 - 8 C. ICM-Pro scores the binding of a ligand to a receptor based upon the comparison of a series of small molecule/ protein interactions that have been reported in the PDB database. A rigid receptor/flexible ligand approach was adopted that uses five potential energy maps combining hydrophobicity, electrostatics, hydrogen bond formation, and two van-der-Waals parameters. In all cases, the program’s default parameters were used.
3. Results and Discussion
The starting material 4,6-dimethyl-1H-pyrazolo[3,4-b] pyridin-3-amine (1) was prepared according to the reported method [19,20], which was alkylated, after its treatment with anhydrous potassium carbonate in dry N,N-dimethylformamide, with ethyl-2-chloroacetate to give Ethyl 2-(3-amino-4,6-dimethyl-1H-pyrazolo[3,4-b] pyridin-1-yl)acetate (2) in good yield. The 1H NMR spectrum of derivative 2 showed that N-CH2 of the N-alkylated product appeared as a triplet at δ 1.13 and quartet at δ 4.22 ppm respectively. The latter derivative 2 was treated with hyfrazine hydrate, in ethanol on boiling, to afford the corresponding acid hydrazid. 2-(3-Amino- 4,6-dimethyl-1H-pyrazolo[3,4-b]pyridin-1-yl) acetohydrazide (3) was cyclized on treating with carbon disulfide, in basic medium, to give the 5-((3-amino-4,6-dimethyl- 1H-pyrazolo[3,4-b]pyridin-1-yl)methyl)-1,3,4-oxadiazole-2-thiol (4) which showed the broad singlet at δ 13.82 ppm corresponding to the SH group in its H1 NMR, the latter oxadiazole 4 was also elucidated via its alkylation reactions with some alkylation agents such as methy, ethyl iodide and benzyl chloride to afford the S-alkyl derivatives 5a-c. These alkylated derivatives were elucidated by the MS spectra which showed the molecular ion peaks corresponding to its molecular wights (Scheme 1).
The acetohydrazide 3 was treated with phenylisothiocyanate to give the corresponding phenylhydrazinecarbothioamide derivative 6. which was elucidated, besides the 1H NMR and IR, by the mass spectrum which showed the molecular ion peak at m/z 270 corresponding to [M+ + 1], m/z When the thiosemicarbazide 6 was reacted with concentrated sulfuric acid. 1,3,4-thiadiazole derivative 7 was obtained which showed in its 1H NMR spectrum of the broadband of the NHPh at δ 11.55 ppm. The formation of the thiadiazole ring, under such acidic conditions, is due to the loss of nucleophilicity of N-4 as a result of its protonation leading to an increase in the nucleophilicity of the sulfur atom toward the attack of the carbonyl carbon as shown in Scheme 2. On the other hand, when the cyclization of 6 was carried out under basic conditions, the nucleophilicity of N-4 was enhanced and affording cyclization with carbonyl carbon atom to afford 1,2,4-triazole derivative in 67% yield. In the treatment of thiosemicarbazide 6 with mercuric oxide, the cyclization was performed, affording the 1,3,4- oxadiazole derivative 9. The method of cyclization includes desulfurization by HgO. The 1H NMR of triazole 8 showed a broad singlet at δ 12.05 ppm corresponding to the SH group. The oxadiazole derivatve 9 was elucidated besides, NMR, elemental analysis, by the mass spectrum which showed a [M+] peak, in agreement with its molecular formula (Scheme 3).
Molecular Modeling Studies
To pre-assess the anti-tumorigenic behavior of our NAlkylated, 1,2,4-triazole, 1,3,4-oxa-, thiadiazole and complexes containing the aminopyrazolopyridine derivatives 5(a-c) and (7-9) on a structural basis, automated docking studies were carried out using MOLSOFT ICM 3.4 - 8C program [21]. The scoring functions and hydrogen bonds formed with the surrounding amino acids are used to predict their binding modes, their binding affinities and orientation of these compounds at the active site of AKR1C3 enzyme. The protein-ligand complex was constructed based on the X-ray structure (PDB entry 1S2A) AKR1C3 with its bound inhibitor indomethacin [22].
The scoring functions of the compounds were calculated from minimized ligand protein complexes. The X-ray crystal structure of AKR1C3 reveals a substratebinding site that consists mainly of: hydrophobic aromatic amino acid side chains (Tyr24, Tyr55, Leu54, Trp227, and Phe306). An oxyanion hole, which is located at the bottom of the hydrophobic pocket, is formed by active site tyrosine (Tyr55), histidine (His117), and the coenzymes nicotinamide ring [21].

Scheme 1.Formation of the oxadiazole derivatives.

Scheme 2.Schematic investigation according to the condition applied.
In our investigation, the 3D-coordinates in X-ray crystal structure of AKR1C3 in complex with the ligand, indomethacin (PDB entry 1S2A) [21] was used as the receptor model in AKR1C3 docking simulation. The docked model of indomethacin with AKR1C3 (Figure 1) was consistent with the previously reported X-ray analysis [22] and revealed the following binding mode: The carbonyl oxygen is far away from Tyr 55 or His 117 to H-bond directly. Instead, the carboxylate group points toward and interacts with the oxygen atoms O1n, O2n from the nicotinamide half of the NADP+ diphosphate moiety, forming two hydrogen bonds, and additional H-bond if formed between indomethacin O2 of COOH and He2 of Glu 222.
Our active compounds 5(a-c) and (7-9) when modeled in the active site of AKR1C3 enzyme (Table 1, Figures 2,3) revealed strong binding affinities. Their binding energies were −67.08, −88.58, −96.73, −103.82, −88.74 and −97.28 Kcal/mol, respectively compared to −80.45 of
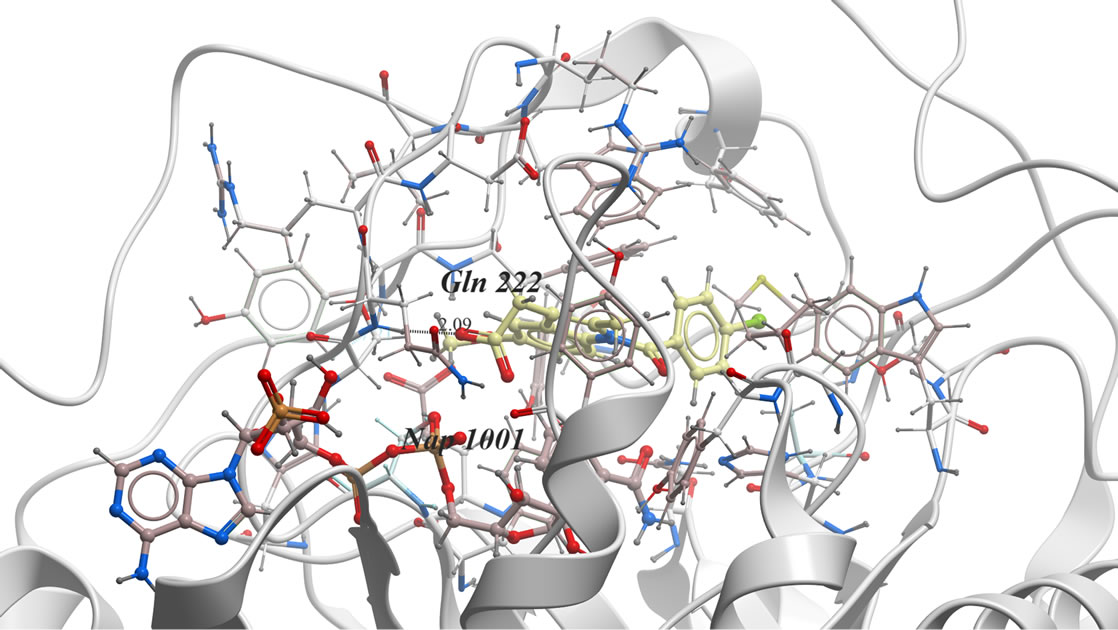
Figure 1. The proposed binding mode of original ligand indomethacin into the active binding site of AKR1C3 active site. It has ICM score of −80.45, it forms three hydrogen bonds, two of them between H of COOH and O1n, O2n of Phosphate moiety of NADP. And another hydrogen bond between O2 of COOH and He2 of amino acid Glu222….
indomethacin. It is interesting to point out that compounds 5(a-c) and (7-9) are found to be very promising


Table 1. Docking of compounds on AKR1C3.

Scheme 3.crystallization of the thiosemicarbazole derivative.

Figure 2. The proposed binding mode of 5a into the active binding site of AKR1C3 active site. It has ICM score of −67.08, it forms three hydrogen bonds, two of them between O2 of OH and He2 and He3 of Tyr24 and Glu58…One hydrogen bond between O3 of SO2 and Hh1 of amino acid Arg 226. And hydrogen bond between O4 of SO2 and H1n1 of Phosphate moiety of NADP….

Figure 3. The proposed binding mode of 5b into the active binding site of AKR1C3 active site. It has ICM score of −88.58, it forms 13 hydrogen bonds, one between O1 of OCH3 and Ha1 of Phosphate moiety of NADP. Two hydrogen bonds between O4 of SO2 and Hd2 of Arg31 and H7n7 of Amide of Nicotinamide ring … three hydrogen bonds, two of them between O2 of OH and He2 and He3 of Tyr24 and Another two hydrogen bonds is between H14, H15 of benzene ring of sulfonamide and He2 of amino acid Tyr55.
AKR1C3 inhibitors, as they take advantageof their ability to make H bond with the amino acid present in the oxyanion hole (Tyr 55 and Glu222) in the presence of the coenzyme’s nicotinamide with the aromatic residues located in the hydrophobic pocket, and at the same time to NADP+ diphosphate.
Using Molsoft ICM 3.4 - 8C program, molecular modelling and docking studies of the synthesized compounds into ARK1C3 complexed with its bound inhibitor indomethacin (1S2A) were performed in order to predict The binding affinities and orientations of these compounds at the active site. The ICM score values of 5(a-c) and (7-9) were −67.08, −88.58, −96.73, −103.82, −88.74 and −97.28 Kcal/mol, respectively compared to −80.45 of indomethacin. These derivatives will encourage researchers to help to design future anticancer agents with therapeutic potentials.
NOTES