Design, Synthesis and Inhibitory Properties against Coxsackie B3/B6 of Some Novel Triazole Derivatives ()
1. Introduction
In recent years heterocyclic compounds analogues and derivatives have gained significant interest in various fields of drug discovery due to their wide range of biological activities, such as, anti-microbial, anti-tumor, anthelmintic, anti-leishmanial, anti-convulsant and antiinflammatory [1]. And triazoles as important aromatic nitrogen-containing heterocycle possess a lot of desirable features and have been incorporated into a wide variety of potent therapeutic agents [2]. Introduction of a triazole ring into nucleosides to improve bioactivity in antivirus agents has become widespread in drug design practices since the first synthetic nucleoside drug, ribavirin, showed a broad spectrum of antiviral activity against many RNA and DNA viruses.
Human enteroviruses are the most common cause for the majority of human respiratory diseases along with rhinoviruses. Moreover, enteroviruses may cause aseptic meningitis, encephalitis, febrile illness, hand-foot-mouth disease, and myocarditis [3]. It is estimated that worldwide enteroviruses cause 10 - 15 billion (or more) enteroviral infections annually, and many outbreaks have been reported from Asian countries [4]. With more than 200 enterovirus serotypes existing, there is no effective antiviral drug for clinical use of enteroviral infections so far [5]. This highlights the urgency and significance for developing anti-enterovirus agents.
Enteroviruses are non-enveloped, single-stranded (+) RNA viruses belonging to the picornavirus family, which associated with several human and mammalian diseases [6]. Conventionally, enteroviruses were classified into polioviruses, coxsackie A viruses (CVA), coxsackie B viruses (CVB), echoviruses, and enteroviruses 68 - 71 (EV 68 - 71). Coxsackie viruses, and in particular CVB 3, have often been associated with the development of myocarditis, which may lead to sudden death in young adults or progress to dilated cardiomyopathy [7,8].
In our previous work [9,10], a series of 2-pyridyl- 1H-benzimidazole-4-carboxamide derivatives were synthesized, screened and indentified as modest inhibitors of CVB3. In view of its novel structural template, which differed from those of all reported anti-enterovirus agents, we were interested to introduce 1,2,4-triazoles moiety. A series of amide derivatives containing 1,2,4-triazole ring were synthesized, screened and indentified as modest inhibitors of coxsackie viruses. It was believed that these compounds could be found effective against picornavirus. The inhibitory activities of these triazole derivatives were tested against CVB3, CVB6. No active clinical drugs against enterovirus employed by now, a relatively effective drug, ribavirin (RBV), was selected as the positive control drug. These triazole derivatives were found to exhibit good inhibitory activities against the two kinds of enteroviruses.
2. Experimental
2.1. General
The synthetic procedures for our desired compounds are outlined in Figure 1. Briefly, the triazoles (1) were synthesized with thiosemicarbazide and carboxylic acid [11], and then reacted with halides using sodium hydroxide as base to produce the key intermediates (2) [12]. Only methyl iodide and 4-chloro-1,3-dinitrobenzene could react with the 1,2,4-triazoles in good yield. With another key intermediate 5-methyl-3-phenylisoxazole-4-carboxylic acid (7) in hands, compounds 3 were obtained by one step with excellent yield of 80% under regular EDC/ HOBt conditions [10]. 3-Chloroperbenzoic acid (MCPBA) was chosen as an oxidant to convert thioethers into sulfones.
The important intermediate 5-methyl-3-phenylisoxazole-4-carboxylic acid (7) was synthesized with benzaldehyde as the starting material (Figure 2). (E)-benzaldehyde oxime (5) was converted into 5-methyl-3-phenylisoxazole-4-carboxylic acid (7) in two steps. A nitrile oxide cycloaddition was employed to generate the trisubstituted isoxazole (6). Then the ester was saponified to yield acid (7). They were used directly without further purification in the next step [13,14].
The virus strains of CVB3, CVB6 were provided by American Type Culture Collection (ATCC). The positive control drug, RBV was produced by Hubei Keyi Pharmaceutical Factory. The newly synthesized triazole compounds (3a-4a) were dissolved in DMSO and diluted with the culture medium. Vero cells were planted in 96-well culture plates. After 24 h the plates were placed in the corresponding virus bulk for 2 h. Then the solutions of triazole compounds and RBV were added in the plates and cell wells and virus wells were set simultaneously. When the cytopathic effect (CPE) of virus wells was over 4, the CPE of cell wells was observed. The concentration required to inhibit virus growth by 50% (IC50) was determined by the Reed-Muench method [15].
2.2. Compound Data
1-(3-(2,4-dinitrophenylthio)-1H-1,2,4-triazol-1-yl)ethanone (3a).
Yield 77.2%. m.p. 151˚C. 1HNMR (DMSO, 400 MHz) δ: 2.8 73 (s, 3H, CH3), 7.131 - 7.154 (d, 1H, ArH), 8.347 - 8.376 (m, 1H, ArH), 8.896 - 8.902 (d, 1H, ArH), 8.958 (s, 1H, ArH). MS m/z 310 [M + H]+. Anal. Calcd for C10H7N5O5S: C, 38.84; H, 2.28; N, 22.65; S, 10.37. Found: C, 38.92; H, 2.07; N, 22.01; S, 11.19.
(3-(2,4-dinitrophenylthio)-1H-1,2,4-triazol-1-yl)(phen yl)methanone (3b).
Yield 57.2%. m.p. 146˚C. 1HNMR (DMSO, 400 MHz) δ: 7.3 78 - 7.483 (m, 5H, ArH), 7.510 - 7.588 (m, 2H, ArH), 7.694 - 7.715 (d, 1H, ArH), 7.957 - 7.978 (d, 1H, ArH). MS m/z 372 [M + H]+. Anal. Calcd for C15H9N5O5S: C, 48.52; H, 2.44; N, 18.86; S, 8.64. Found: C, 48.83; H, 2.08; N, 18.22; S, 9.32.
(3-(2,4-dinitrophenylthio)-1H-1,2,4-triazol-1-yl)(5-
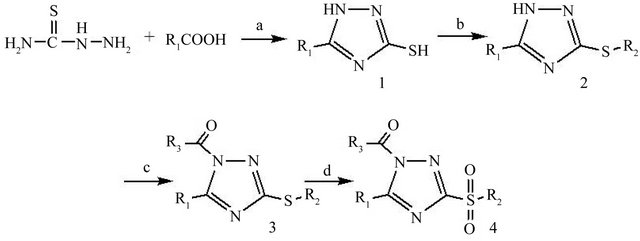
Figure 1. Reagents and conditions: (a) toluene, reflux; (b) Halides, NaOH, CH3CH2OH, rt; (c) R2COOH, EDC/HOBt, Et3N, CH2Cl2, rt; (d) MCPBA, CH2Cl2, 0˚C→rt.
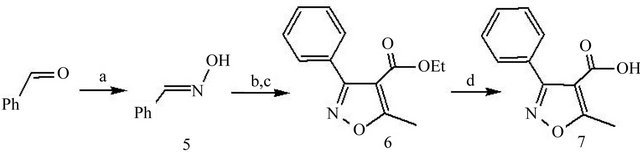
Figure 2. Reagents and conditions: (a) NH2OH∙HCl/Na2CO3, CH3CH2OH, rt; (b) NCS, DMF, rt; (c) CH3CH2ONa/EAA, CH3CH2OH, 50˚C; (d) NaOH, CH3CH2OH/H2O, rt.
methyl-3-phenylisoxazol-4-yl)methanone (3c).
Yield 66.3%. m.p. 138˚C. 1HNMR (DMSO, 400 MHz) δ: 2.6 73 (s, 3H, CH3), 7.378 - 7.416 (m, 1H, ArH), 7.448 - 7.467 (d, 2H, ArH), 7.510 - 7.548 (m, 1H, ArH), 7.569 - 7.588 (m, 2H, ArH), 7.694 - 7.715 (d, 1H, ArH), 7.795 - 7.978 (d, 1H, ArH). MS m/z 453 [M + H]+. Anal. Calcd for C19H12N6O6S: C, 50.44; H, 2.67; N, 18.58; S, 7.09. Found: C, 50.71; H, 2.83; N, 18.17; S, 8.35.
1-(3-(2,4-dinitrophenylthio)-5-(trifluoromethyl)-1H-1,2,4-triazol-1-yl)ethanone (3d).
Yield 54.5%. m.p. 154˚C. 1HNMR (DMSO, 400 MHz) δ: 2.6 48 (s, 3H, CH3), 7.539 - 7.587 (m, 1H, ArH), 8.026 - 8.044 (m, 1H, ArH), 8.224 (s, 1H, ArH). MS m/z 378 [M + H]+. Anal. Calcd for C11H6F3N5O5S: C, 35.02; H, 1.60; N, 18.56; S, 8.50. Found: C, 35.63; H, 2.13; N, 18.22; S, 8.97.
(3-(2,4-dinitrophenylthio)-5-(trifluoromethyl)-1H-1,2,4-triazol-1-yl)(phenyl)methanone (3e).
Yield 34.2%. m.p. 150˚C. 1HNMR (DMSO, 400 MHz) δ: 7.4 82 - 7.511 (m, 4H, ArH), 7.571 - 7.590 (m, 1H, ArH), 7.654 - 7.675 (m, 1H, ArH), 8.459 - 8.490 (m, 1H, ArH), 8.745 - 8.737 (d, 1H, ArH). MS m/z 440 [M + H]+. Anal. Calcd for C16H8F3N5O5S: C, 43.74; H, 1.84; N, 15.94; S, 7.30. Found: C, 44.12; H, 2.04; N, 15.28; S, 8.37.
(3-(2,4-dinitrophenylthio)-5-(trifluoromethyl)-1H-1,2,4-triazol-1-yl)(5-methyl-3-phenylisoxazol-4-yl)methanone (3f).
Yield 54.3%. m.p. 144˚C. 1HNMR (DMSO, 400 MHz) δ: 2.6 73 (s, 3H, CH3), 7.378 - 7.510 (m, 5H, ArH), 7.571 - 7.586 (m, 1H, ArH), 7.695 - 7.715 (d, 1H, ArH), 7.797 - 7.978 (d, 1H, ArH). MS m/z 521 [M + H]+. Anal. Calcd for C20H11F3N6O6S: C, 46.16; H, 2.13; N, 16.15; S, 6.16. Found: C, 46.45; H, 2.01; N, 15.87; S, 7.08.
1-(3-(2,4-dinitrophenylthio)-5-phenyl-1H-1,2,4-triazol-1-yl)ethanone (3g).
Yield 47.5% .m.p.167˚C. 1HNMR (DMSO, 400 MHz) δ: 2.7 34 (s, 3H, CH3), 7.472 - 7.557 (m, 5H, ArH), 8.357 - 8.385 (d, 1H, ArH), 8.552 - 8.581 (m, 1H, ArH), 8.902 - 8.928 (d, 1H, ArH). MS m/z 386 [M + H]+. Anal. Calcd for C16H11N5O5S: C, 49.87; H, 2.88; N, 18.17; S, 8.32. Found: C, 49.98; H, 2.56; N, 17.75; S, 9.04.
(3-(2,4-dinitrophenylthio)-5-phenyl-1H-1,2,4-triazol-1-yl)(phenyl)methanone (3h).
Yield 46.2%. m.p. 153˚C. 1HNMR (DMSO, 400 MHz) δ: 7.4 73 - 7.510 (m, 4H, ArH), 7.538 - 7.557 (t, 1H, ArH), 7.771 - 7.766 (m, 2H, ArH), 7.881 - 7.905 (m, 2H, ArH), 8.276 - 8.297 (d, 1H, ArH), 8.356 - 8.386 (d, 1H, ArH), 8.552 - 8.580 (m, 1H, ArH), 8.904 - 8.928 (d, 1H, ArH). MS m/z 448 [M + H]+. Anal. Calcd for C21H13N5O5S: C, 56.37; H, 2.93; N, 15.65; S, 7.17. Found: C, 56.86; H, 2.53; N, 14.83; S, 7.63.
(3-(2,4-dinitrophenylthio)-5-phenyl-1H-1,2,4-triazol-1- yl)(5-methyl-3-phenylisoxazol-4-yl)methanone (3i).
Yield 31.3%. m.p.145˚C. 1HNMR (DMSO, 400 MHz) δ: 2.7 18 (s, 3H, CH3), 7.472 - 7.510 (m, 5H, ArH), 7.535 - 7.557 (m, 1H, ArH), 7.721 - 7.766 (m, 2H, ArH), 7.881 - 7.905 (m, 2H, ArH), 8.276 - 8.297 (d, 1H, ArH), 8.356 - 8.385 (q, 1H, ArH), 8.553 - 8.580 (q, 1H, ArH). MS m/z 529 [M + H]+. Anal. Calcd for C25H16N6O6S: C, 56.82; H, 3.05; N, 15.90; S, 6.07. Found: C, 57.02; H, 3.21; N, 15.68; S, 6.64.
1-(3-(methylthio)-1H-1,2,4-triazol-1-yl)ethanone (3j).
Yield 34.5%. m.p. 175˚C. 1HNMR (DMSO, 400 MHz) δ: 2.7 18 (s, 3H, CH3), 2.734 (s, 3H, CH3), 8.904 - 8.922 (m, 1H, ArH). MS m/z 158 [M + H]+. Anal. Calcd for C5H7N3OS: C, 38.20; H, 4.49; N, 26.73; S, 20.40. Found: C, 38.33; H, 4.05; N, 25.95; S, 21.38.
(3-(methylthio)-1H-1,2,4-triazol-1-yl)(phenyl)methanone (3k).
Yield 36.2%. m.p. 169˚C. 1HNMR (DMSO, 400 MHz) δ: 2.7 34 (s, 3H, CH3), 7.473 - 7.557 (m, 5H, ArH), 8.911 - 8.928 (m, 1H, ArH). MS m/z 220 [M + H]+. Anal. Calcd for C10H9N3 OS: C, 54.78; H, 4.14; N, 19.16; S, 14.62. Found: C, 55.14; H, 4.01; N, 18.75; S, 15.38.
(5-methyl-3-phenylisoxazol-4-yl)(3-(methylthio)-1H-1, 2,4-triazol-1-yl)methanone (3l).
Yield 56.1%. m.p. 165˚C. 1HNMR (DMSO, 400 MHz) δ: 2.7 18 (s, 3H, CH3), 2.734 (s, 3H, CH3), 7.473 - 7.510 (m, 5H, ArH), 8.904 - 8.922 (m, 1H, ArH). MS m/z 301 [M + H]+. Anal. Calcd for C14H12N4O2S: C, 55.99; H, 4.03; N, 18.65; S, 10.68. Found: C, 56.33; H, 4.35; N, 18.29; S, 11.32.
(3-(2,4-dinitrophenylsulfonyl)-1H-1,2,4-triazol-1-yl)(5- methyl-3-phenylisoxazol-4-yl)methanone (4a).
Yield 25.1%. m.p. 159˚C. 1HNMR (DMSO, 400 MHz) δ: 2.6 73 (s, 3H, CH3), 7.378 - 7.548 (m, 5H, ArH), 7.564 - 7.588 (m, 2H, ArH), 7.694 - 7.715 (d, 1H, ArH). MS m/z 485 [M + H]+. Anal. Calcd for C19H12N6O8S: C, 47.11; H, 2.50; N, 17.35; S, 6.62. Found: C, 47.43; H, 2.32; N, 17.08; S, 7.57.
3. Results and Discussion
The cytotoxicity and antiviral potency of these synthesized 1,2,4-triazole derivatives were evaluated in vero cells against CVB3/B6. No active clinical drugs against CVB3/B6 employed by now, RBV was the recommended clinical antiviral drug in China, the IC50 values of RVB were provided as comparable data.
The results were summarized in Table 1. The antienterovirus activity of each compound was expressed as the concentration of compound that achieved 50% inhibition (IC50) of enterovirus growth. The cytotoxicity of each compound was expressed as the concentration of compound required to kill 50% (TC50) of the vero cells. As a major pharmaceutical parameter for possible future clinical development, the selectivity index (SI) was de-
Table 1. Activity of the triazole derivatives against coxsackie virus B3/B6 in vero cells.

aCytotoxic concentration required to inhibit vero cell growth by 50%; bConcentration required to inhibit CVB3 growth by 50%; cSelectivity index values equaled to TC50/IC50; dNot active in the largest concentration which were not toxic to vero cells.
termined as the ratio of TC50 to IC50. The bioactivity of each compound was evaluated by the combination of its IC50 and SI.
As shown in Table 1, the 1,2,4-triazole derivatives (3a-4a) were evaluated against CVB3. It is found that most of the synthesized compounds had good antiviral activity, far more active than RVB with IC50 value of 384.90μM. Each of the bioactive compounds possessed an IC50 value within the range from 1.71 μM to 66.67 μM, and a TC50 value ranging from 4.14 μM to more than 200 μM. Analyzing the activities of the synthesized compounds, the following structure-activity relationship (SAR) was observed.
In series 3a-l, IC50s of compounds 3a and 3b (IC50 = 2.47 and 2.47 μM, respectively) were better than compounds 3d and 3e (IC50 = 22.22 and 38.49 μM, respectively). However, compounds 3a and 3b had pronounced cytotoxicity (TC50 = 7.41 and 7.41 μM, respectively) resulting in similar selective indices of compounds 3d and 3e. It was indicated that the antiviral activity could be reduced and the cytotoxicity could be enhanced due to the introduction of trifluoromethyl in the R1 position. Compound 3g with phenyl at the R1 position showed higher IC50 (IC50 = 1.71 μM) than compound 3a (IC50 = 2.47 μM), but it appeared to be more toxic (TC50 = 4.14 μM) than compound 3a (TC50 = 7.41 μM). Compound 3h with phenyl at the R1 position showed much lower IC50 (IC50 = 66.67 μM) than compound 3b (IC50 = 2.47 μM), but it appeared to be less toxic (TC50 = 138.67 μM) than compound 3b (TC50 = 7.41 μM). Generally, the antiviral regularity of this series was inconspicuous.
Compared with compound 3g (IC50 = 1.71 μM), all other derivatives showed equivalent or decreased antiviral activities. Due to the introduction of dinitro phenyl in the R2 position, compounds 3a-h exhibited good antiviral potency against CVB3.The introduction of methyl in the R2 position resulted in approximately no biological activities, but their cytotoxicities were significantly decreased with TC50 values of more than 200 μM. Compounds 3j-k (IC50 > 66.67 μM) with methyl substituent at the R2 position caused significantly decrease in antiviral activity, but showed lower cytotoxicity with TC50 values of more than 200 μM.
Compound 3c (IC50 = 22.22 μM) with moiety of isoxazole at the R3 position displayed lower activity than compounds 3a and 3b (IC50 = 2.47 and 2.47 μM, respectively), but was less cytotoxic with TC50 value of 96.15 μM. Compound 3d with methyl at the R3 position showed higher IC50 (IC50 = 22.22 μM) than compound 3e with phenyl at the R3 position (IC50 = 38.49 μM). Compound 4a proved inactive against CVB3/B6 in vero cells which was the oxidation product of compound 3a, but the cytotoxicity to vero cells was decreased.
These structure-activity relationship analyses indicated that the introduction of dinitro phenyl in the R2 position was critical for the high antiviral activity, such as 3a, 3b, 3g. The bioactive compounds 3a, 3b, 3g expressed better IC50s (IC50 = 2.47, 2.47, 1.71μM). They could be considered as promising candidates for the development of new derivatives with anti-enterovirus activity and for additional studies concerning the antiviral activity of this group of compounds. The other bioactive compounds 3cf, 3h showed higher IC50s (IC50 > 20 μM), but they appeared to be less toxic (TC50 = 96.15, 66.67, 138.67, 46.22, 138.67 μM, respectively).
Compounds 3a-4a were screened against CVB6 (Table 1).There were only four compounds 3a, 3g, 3h, 3l shown good antiviral potency against CVB6 (IC50 = 1.43, 1.43, 28.64 and 22.22 μM, respectively).The most selective compound against CVB6 was compound 3a, with SI of 5.2, IC50 of 1.43 μM, TC50 of 7.41 μM. Comparing these results with those shown in Table 1, it was found that the similarity existed among antiviral activities against CVB3,CVB6 of certain compounds (3a, 3g, 3h), indicating that a compound seemed to be effective against other virus if it was effective against one virus of the same family. Compound 3a (IC50 = 2.47 and 1.43 μM, respectively) and 3g (IC50 = 1.71 and 1.43 μM, respectively) exhibited good antiviral activities against both CVB3 and CVB6, but performed toxic (TC50 < 10 μM). Compound 3h displayed lower cytotoxicity to vero cells (TC50 > 100 μM).
4. Conclusion
In summary, a novel series of 1,2,4-triazole derivatives (3a-4a) have been synthesized and evaluated for their antiviral activities against CVB3, CVB6 replication in cell culture. Most of the 1,2,4-triazole derivatives pronounced good anti-CVB3 activities and some pronounced good anti-CVB6 activities. Moreover, some of the synthesized compounds had excellent antiviral activity, but they were cytotoxic to cells, such as compounds 3a, 3b and 3g (IC50 < 3 μM, TC50 < 10 μM). And some of the synthesized compounds had low cytotoxicities, such as 3h (IC50 > 20 μM, TC50 > 100 μM), but their antiviral activites were not eminent. Further studies are in progress in our laboratories to increase the antiviral potency and decrease cytotoxicity, most importantly, to improve the TC50/IC50 ratio. The modifications on 1,2,4- triazoles moiety display valuable biological activities, and these modifications can be utilized as potent therapeutic agents in future.
5. Acknowledgements
We thank Ms. Chen Yan for 1HNMR support.
NOTES