1. Introduction
β-Lactams, being a structural unit found in the most widely used antibiotics [1], have occupied a basic position in medicinal chemistry for almost a century now. With the microbes basic position in medicinal chemistry for almost a century now. With the microbes responding to the traditional antibiotics through β-lactamases, the need for novel antibiotics prevails, making synthesis of newer β-lactams ever more important. In addition to their use as antibiotics, β-lactams are increasingly being used as synthons for other biologically important molecules [2-11]. β-Lactams have been found to act as cholesterol acyl transferase inhibitors [12], thrombin inhibitors [13], human cytomegalovirus protease inhibitors [14], matrix metalloprotease inhibitors [15], cysteine protease [16], and apoptosis inductors [17]. Spirocyclic β-lactams have attracted attention as they have been shown to be β-turn mimetics [18,19] and precursors for α,α-disubstituted β-amino acids [20]. The chartelline has a spiro-β-lactam moiety in its structure marine natural products [21]. It has been found that spiro-β-lactams act as poliovirus and human rhinovirus 3C-proteinases inhibitors [22]. These compounds are mostly synthesized by cycloaddition to an exocyclic bond. Several syntheses of spiro-β-lactams have been reported [23-42]. Polycyclic aromatic β-lactams have shown anticancer and other biological activities [43-45].
2. Experimental
2.1. General
Melting points are uncorrected. IR spectra were measured as KBr pelltes on a pye-unicam sp 1000 spectrophotometer. 1H-NMR spectra were recorded in [2H6] dimethyl sulfoxide at 200 MHz on a Varian Gemini NMR spectrometer using, MeSi as internal reference. Mass spectra were obtained on a shimadzu GCMS-QP 1000EX mass spectrometer at 70 eV. Elemental analysis was carried out at the Microanalytical Center of Cairo University.
2.2. Synthesis of New 4,9-Diamino-2,3,7,8- tetramethyl-1,5a,6,10a-tetrahydropyrido [2,3-g]quinoline-5,10-dione1
A solution of the appropriate acetamide (0.59 g, 0.02 mole) and malononitrile (0.66 g, 0.02 mole) which prepared in situ in ethanol (30 ml) containing (0.5 ml) piperidine was treated with 1,4-p-benzoquinone (0.02 mole, 1.58 g). The reaction mixture was heated under reflux for 13 - 15 h (monitored by TLC). The solvent was then evaporated under reduced pressure. Poured onto ice/ water acidified by HCl, the solid product so formed was collected by filtration and crystallized from ethanol, yield 75%; Mp: 295˚C; IR (KBr, Cm−1): ν~ 3100 - 3400 (NH, NH2), 2214 (2CN), 1665 (2C=O); 1H-NMR δ (ppm): 1.63 (d. S, 6H, 2CH3), 6.01 (brs, 4H, 2NH2), δ 7.01 - 8.01 (m, 2H, Ar-H), δ 9.67 (brs, 2H, 2NH); 13C-NMR δ (ppm): 174.09 (C=O), 168.4 (C=O), 152.5 (carbon which attach by CH3), 119.4 (carbon which attach by CN), 136.5 (carbon which attach by NH2) 98 - 135 (C=C), 63 (C-N), 24 (CH3), 117.3 (CN); MS (334); analysis calculatedfor C17H14O2N6: C, 61.0; H, 4.22; N, 25.22%. Found: C, 59.95; H, 4.02; N, 25.01%.
2.3. Synthesis of New 4,9-diamino-2,7-dimetyl- 5,10-dioxo-1,5,5a,6,10,10-ahexahydropyrido- [2,3-g]quinoline-3,8-dicarboxamide-2
A mixture of compound 1 (3.34 g, 0.01 mole) and conc. Sulfuric acid (20 ml) was stirred at room temperature. The reaction mixture was filtred and crystallized from ethanol to give compound 2 yield 73%; Mp: 285˚C; IR (KBr, Cm−1): ν~ 3150 - 3400 (NH, NH2), 1670 (2CO), 1645 (2CO); 1H-NMR δ (ppm): δ 1.64 (d. S, 6H, 2CH3), 6.49 (brs, 4H, 2NH2), δ 7.01 - 8.01 (m, 6H, Ar-H), δ 10.50 (brs, 2H, 2NH). 13C-NMR δ (ppm): 173.5 (C=O), 167.5 (C=O), 165.5 (C=O which attach by NH2), 153.5 (carbon which attach by CH3), 120.4 (carbon which attach by CONH2), 137 (carbon which attach by NH2) 98 - 135 (C=C), 64 (C-N), 23.5 (CH3); MS (370); analysis calculated for: C17H18O4N6: C, 55.08; H, 4.89; N, 22.77%. Found: C, 55.01; H, 4.93; N, 22.03%.
2.4. Synthesis of New Compound 4
A mixture of compound 2 (3.70 g, 0.01 mole) and sulfur (0.32 g, 0.01 mole) in pyridine 30 ml was refluxed for 15 h. The reaction mixture was filtred hot from the unreacted material, the filtrate was triturated with ice water acidified by conc. Hydrochloric acid the precipitate filtred, and crystallized from the proper solvent to give compound 4 Yield 76%; Mp > 300˚C; IR (KBr, Cm−1): ν~ 3100 - 3400 (NH, NH2), 1670 (2CO), 1645 (2CO); 1HNMR δ (ppm): δ 6.02 (brs, 4H, 2NH2), δ 7.01 - 8.01 (m, 6H, Ar-H), δ 9.51 (brs, 2H, 2NH); 13C-NMR δ (ppm): 173.5 (C=O), 167.5 (C=O), 164.5 (C=O which attach by NH2), 120.4 (carbon which attach by CONH2), 137.5 (carbon which attach by NH2) 98 - 135 (C=C), 63.5 (C-N), 32 (CH2S); MS (430); analysis calculated for: C17H14O4N6S2: C, 47.44; H, 3.28; N, 19.52; S, 14.89%. Found: C, 47.21; H, 3.36; N, 19.35; S, 14.81%.
2.5. Synthesis of New Schiff Bases 5a-c
To a stirred solution of compound 4 (4.30 g, 0.01 mole) in a mixture of ethanol (20 ml) and DMF (10 ml), different aromatic amine (2.22 g, 0.02 mole; 2.44 g, 0.02 mole; 3.02 g, 0.02 mole) respectively in presence of (0.5 ml) piperidine catalyst. The reaction mixture was heated under reflux for 9 h. The reaction mixture diluted with ice water and neutralized with hydrochloric acid. The solid product formed on standing over night was filtred, washed thoroughly with cold water, and dried, recrystallization from dimethylformamide to give 5a-c.
5a: yield 70%; Mp > 300˚C; IR (KBr, Cm−1) ν~: 3100 - 3400 (NH, NH2), 1680 (2CO), 1586 (C=N); 1H-NMR δ (ppm): 6.69 (brs, 4H, 2NH2), δ 7.01 - 8.01 (m, 16H, Ar-H), δ 10.46 (brs, 2H, 2NH), δ 11.42 (brs, 2H, 2NH); MS (612); analysis calculated for: C29H24O4N8S2: C, 56.85; H, 3.95; N, 18.29; S, 10.46%. Found: C, 56.80; H, 3.69; N, 18.01; S, 10.25%.
5b: yield 72%; Mp > 300˚C; IR (KBr, Cm−1) ν~: 3100 - 3400 (NH, NH2, OH), 1687 (2CO), 1590 (C=N); 1HNMR δ (ppm): 6.58 (brs, 4H, 2NH2), 7.01 - 8.01 (m, 16H, Ar-H), δ 10.38 (brs, 2H, 2NH), δ 11.26 (brs, 2H, 2NH); MS (644); analysis calculated for: C29H24O6N8S2: C, 54.03; H, 3.76; N, 17.38; S, 9.95%. Found: C, 53.99; H, 3.37; N, 17.26; S, 9.24%.
5c: yield 75%; Mp > 300˚C; IR (KBr, Cm−1) ν~: 3100 - 3400 (NH, NH2), 1690 (2CO), 1593 (C=N); 1H-NMR δ (ppm): 6.71 (brs, 4H, 2NH2), 7.01 - 8.01 (m, 14H, Ar-H), δ 10.51 (brs, 2H, 2NH), δ 11.46 (brs, 2H, 2NH); MS (702); analysis calculated for: C29H22O8N10S2: C, 49.57; H, 3.16; N, 19.93; S, 9.13%. Found: C, 49.52; H, 3.02; N, 19.27; S, 9.01%.
2.6. Synthesis of New β-Lactam 6a-c
A mixture of 5a-c (6.12 g, 6.44 g, 7.02 g, 0.01 mol) respectively and chloroacetyl chloride (1.13 g, 0.02 mole) in a mixture of ethanol (20 ml) and DMF (10 ml) in the presence of (0.5 ml) of triethylamine was refluxed for 15 h. After removal the solvent under reduced pressure, the resulting solid product was filtred and washed with water. The crude product was recrystallized from dimethylformamide and ethanol to yield the corresponding 6a-c.
6a: yield 67% ; Mp > 300˚C; IR(KBr, Cm−1) ν~: 3100 - 3400 (NH, NH2), 1750 (2CO Of βlactam), 1657 (2CO); 1H-NMR δ (ppm): 6.55 (brs, 4H, 2NH2), δ 7.01 - 8.01 (m, 18H, Ar-H), δ 10.39 (brs, 2H, 2NH), 11.44 (brs, 2H, 2NH); 13C-NMR δ (ppm): Aromatic carbon 114 - 157, 163.5 (CO of β lactam); MS (765); analysis calculated for: C33H26O6N8Cl2S2: C, 51.77; H, 3.42; N, 14.63; S, 8.37; Cl, 9.26%. Found: C, 51.59; H, 3.39; N, 14.28; S, 8.19; Cl, 9.15%.
6b: yield 65%; Mp > 300˚C; IR (KBr, Cm−1) ν~: 3150 - 3400 (NH, NH2, OH), 1755 (2CO Of βlactam), 1650 (2CO); 1H-NMR δ (ppm): 6.53 (brs, 4H, 2NH2), δ 7.01 - 8.01 (m, 18H, Ar-H), δ 10.29 (brs, 2H, 2NH), 11.41 (brs, 2H, 2NH); 13C-NMR δ (ppm): 114.5 - 158 (aromatic carbon), 165 (CO of β lactam); MS (797); analysis calculated for: C33H26O8N8Cl2S2: C, 49.69; H, 3.29; N, 14.05; S, 8.04; Cl, 8.89%. Found: C, 49.45; H, 3.16; N, 13.96; S, 7.97; Cl, 8.74%.
6c: yield 69%; Mp > 300˚C; IR (KBr, Cm−1) ν~: 3100 - 3400 (NH, NH2), 1758 (2CO Of β lactam), 1660 (2CO); 1H-NMR δ (ppm): 6.60 (brs, 4H, 2NH2), δ 7.01 - 8.01 (m, 16H, Ar-H), δ 10.43 (brs, 2H, 2NH), 11.50 (brs, 2H, 2NH); 13C-NMR δ (ppm): 115 - 158.5 (aromatic carbon) 165.5 (CO of β lactam); MS (855); analysis calculated for: C33H24O10N10Cl2S2: C, 46.32; H, 2.83; N, 16.37; S, 7.49; Cl, 8.29%. Found: C, 46.04; H, 2.62; N, 16.26; S, 7.37; Cl, 8.13%.
2.7. Synthesis of New Thiazolidinone 7a-c
A mixture of 5a-c (6.12 g, 6.44 g, 7.02 g, 0.01 mole) respectively and mercaptoacetic acid (0.92 g, 0.02 mole) was refluxed in a mixture of ethanol (20 ml) and DMF (10 ml) in the presence of (0.5 ml) of triethylamine was refluxed for 16 h. The resulting residue was triturated with ice water neutralized by concentrated hydrochloric acid and was allowed to stand over night. The solid thus obtained washed with water, dried, and recrystallized from ethanol to yield the corresponding 7a-c.
7a: yield 68%; Mp > 300˚C; IR (KBr, Cm−1) ν~: 3100 - 3400 (NH, NH2), 1700 (2CO), 1655 (2CO); 1H-NMR δ (ppm): 2.51 (d. S, 4H, 2CH2 of thiazolodinone) δ 6.65 (brs, 4H, 2NH2), δ 7.01 - 8.01 (m, 16H, Ar-H), δ 10.47 (brs, 2H, 2NH), 11.35 (brs, 2H, 2NH); MS (760); analysis calculated for: C33H28O6N8S4; C, 52.09; H, 3.71; N, 14.73; S, 16.85%. Found: C, 52.01; H, 3.74; N, 14.45; S, 16.65%.
7b: yield 65%; Mp > 300˚C; IR (KBr, Cm−1) ν~: 3100 - 3400 (NH, NH2, OH), 1692 (2CO), 1650 (2CO); 1HNMR δ (ppm): 2.50 (d. S, 4H, 2CH2 of thiazolidinone), δ 6.62 (brs, 4H, 2NH2), δ 7.01 - 8.01 (m, 16H, Ar-H), δ 10.38 (brs, 2H, 2NH), 11.27 (brs, 2H, 2NH); MS (792); analysis calculated for: C33H28O8N8S4;C, 49.99; H, 3.56; N, 14.13; S, 16.17%. Found: C, 49.86; H, 3.37; N, 14.03; S, 16.01%.
7c: yield 63%; Mp > 300˚C; IR (KBr, Cm−1) ν~: 3150 - 3400 (NH, NH2, OH), 1705 (2CO), 1660 (2CO); 1HNMR δ (ppm): 2.52 (d. S, 4H, 2CH2 of thiazolidinone), δ 6.69 (brs, 4H, 2NH2), δ 7.01 - 8.01 (m, 14H, Ar-H), δ 10.51 (brs, 2H, 2NH), 11.39 (brs, 2H, 2NH); MS (850); analysis calculated for: C33H26O10N10S4: C, 46.58; H, 3.08; N, 16.46; S, 15.07%. Found: C, 46.39; H, 3.02; N, 16.28; S, 14.99%.
3. Result and Discussion
3.1. Synthesis
Our initial strategy in this research project for the synthesis of different structural formulas corresponding spiro systems depended upon the synthesis of the new fused compound 1, which was synthesized by a cycloaddition reaction of equimolecular ratios of the appropriate acetamide and malononitrile with 2 mole of p-benzoquinone (one mole used as an oxidizing agent), in ethanol containing piperidine as catalyst [46]. The cycloaddition reaction leads to the formation of compound 1 through the electronic cyclization according to the suggested mechanism (Scheme 1).
The structure of the new synthesized compound 1 IR spectra which revealed a two carbonyl groups at 1665 cm−1, two cyano group at 2214 cm−1 and 1H-NMR spectra, which revealed the presence of two NH groups at δ 9.67, two NH2 groups at δ 6.01, doublet singlet at 1.63 assigned for two methyl group. The mass spectrum showed the molecular ion peak (M+, C17H14O2N5) at m/z 334. 13C-NMR δ (ppm): 174.09 (C=O), 168.4 (C=O), 152.5 (carbon which attach by CH3), 119.4 (carbon which attach by CN), 136.5 (carbon which attach by NH2) 98 - 135 (C=C), 63 (C-N), 24 (CH3), 117.3 (CN).
When a mixture of compound 1 and conc. Sulfuric acid was stirred at room temperature, hydrolysis of the cyano group took place to give compound 2. The structure of the new synthesized compound 2 was confirmed by its elemental analysis and IR spectra which revealed a carbonyl group at 1645 cm−1 and two carbonyl group at 1665.5 cm−1 and the 1H-NMR spectra, which revealed the presence of two NH groups at δ 10.50, two NH2 group at δ 6.49 doublet, singlet at δ 1.64 assigned for two methyl group. The mass spectrum showed the molecular ion peak (M+, C17H18O4N6) at m/z 370. 13C-NMR δ (ppm): 173.5 (C=O), 167.5 (C=O), 165.5 (C=O which attach by NH2), 153.5 (carbon which attach by CH3), 120.4 (carbon which attach by CONH2), 137 (carbon which attach by NH2) 98 - 135 (C=C), 64 (C-N), 23.5 (CH3). The intelligible bases of IR spectra led us to decide that the isolable structural formula produced from the reaction of compound 2 with sulfuric acid is the structural formula of compound 2 because the absence of peak at 2214 assigned for the cyano group. On the other hand, compound 2 was reacted with sulfur in pyridine which refluxed for 19h to give new compound 4. The structure of the new synthesized compound 4 was confirmed by its elemental analysis and IR spectra which revealed the presence of two carbonyl group of quinone at 1675 cm−1 and two carbonyl group at 1640 cm−1. The 1H-NMR spectra revealed the presence of (brs, 2H, 2NH) at δ 9.51 and (brs, 2H, 2NH) at δ 10.67, (brs, 4H, 2NH2) at δ 6.02 the absence of doublet singlet at δ 1.64 led us to decide that the structural formula of compound 4. The mass spectra showed the molecular ion peak (M+, C17H14O4N6S2) at m/z 430. and 13C-NMR δ(ppm): 173.5 (C=O), 167.5 (C=O), 164.5 (C=O which attach by NH2), 120.4 (carbon which attach by CONH2), 137.5 (carbon which attach by NH2) 98 - 135 (C=C), 63.5 (C-N), 32 (CH2S).
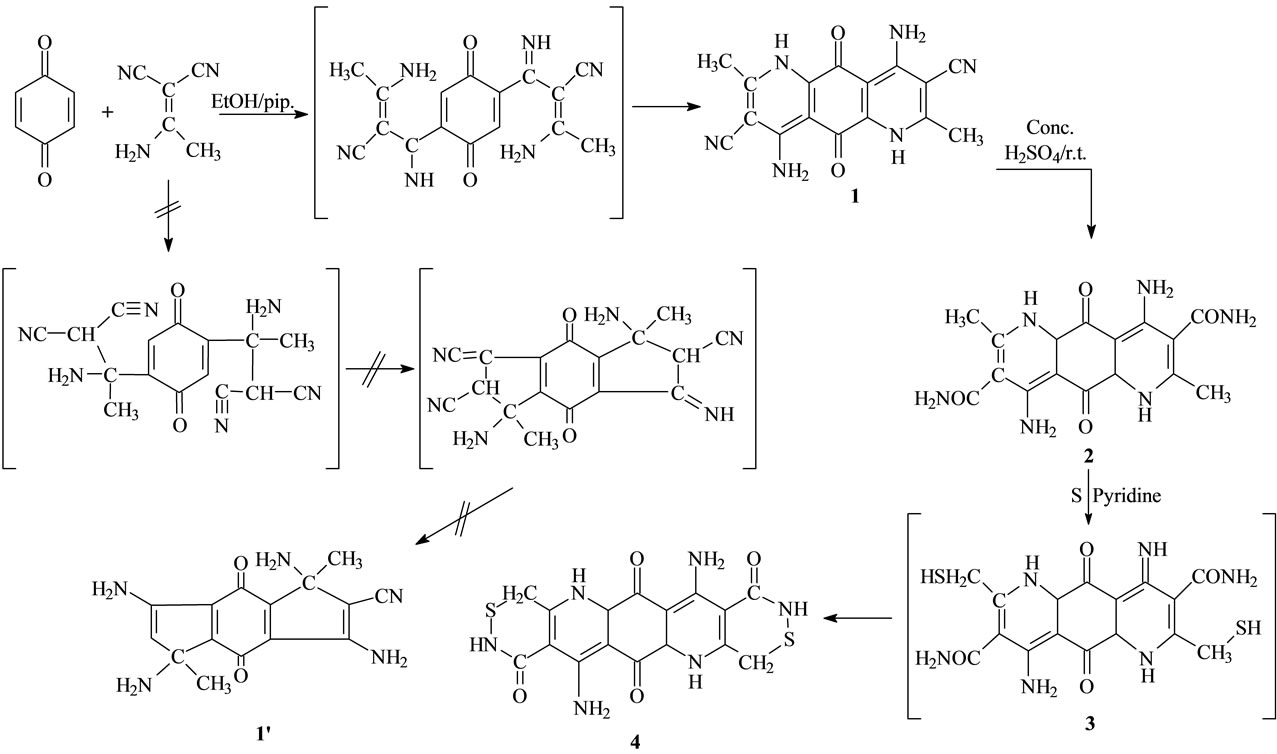
Scheme 1. Illustrate the formation of compound 1, 2 and 4.
It is very important to know that the formation of Schiff bases corresponding to the newly heterocyclic compounds is the cornerstone in the synthesis of the corresponding spiro β-Lactams and thiazolidinone compounds. The activity of the two carbonyl group in compound 4 render compound 4 to react with different aromatic amine in the presence of a mixture of ethanol (20 ml) and DMF (10 ml) as solvent at (0.5 ml) piperidine catalyst to give new Schiff bases 5a-c. The structure of these newly synthesized Schiff base was confirmed by their elemental analysis, IR, 1H-NMR and mass spectra. The activity of azamethine centre in compound 5a-c is more available than the activity of the NH group toward the addition process of chloroacetyl chloride, and this mentioned phenomena is due to the presence of the π electron, which makes the foundation of the δ positive and δ negative charge on the carbon and nitrogen atom, respectively, more easy than the presence of this phenomena on the NH group in which the bonding between nitrogen and hydrogen wheather strong according to the nature of this bonding which leads to decreasing of the mobility desire of the hydrogen atom of this NH group [46]. Thus compound 5a-c reacted with chloroacetyl chloride or mercaptoacetic acid [47] to give spiro β- Lactams and spiro thiazolidinone compounds 6a-c and 7a-c (Scheme 2). The structures of these spiro compounds 6a-c and 7a-c were confirmed by their elemental analysis, IR, 1H-NMR, mass spectra and 13C-NMR.
3.2. 1HNMR and IR
Figure 1 illustrates 1HNMR spectra of compound 1which revealed the presence of two NH groups at δ 9.67, two NH2 groups at δ 6.01, doublet singlet at 1.63 assigned for two methyl group.
Also, Figure 2 indicates 1HNMR spectra of compound 2 revealed the presence of δ 1.64 (d. S, 6H, 2CH3), 6.49 (brs, 4H, 2NH2), δ 7.01 - 8.01 (m, 6H, Ar-H), δ 10.50 (brs, 2H, 2NH). Figure 3 indicates 1HNMR spectra of compound 4 which revealed the absence of absorbtion of two methyl groups; 1H-NMR δ (ppm): δ 6.02 (brs, 4H, 2NH2), δ 7.01 - 8.01 (m, 6H, Ar-H), δ 9.51 (brs, 2H, 2NH).
Figure 4 indicates IR spectra of compound 1:IR(KBr, Cm−1): ν~ 3100 - 3400 (NH, NH2), 2214 (2CN), 1665 (2C=O), which reveald the presence of two cyano groups at 2216 Cm−1 while Figure 5 shows IR spectra of compound 2which indicate the absence of two cyano groups due to hydrolysis process, Figure 6 shows IR spectra of compound 4 as follow, IR (KBr, Cm−1): ν~ 3100 - 3400 (NH, NH2), 1670 (2CO), 1645 (2CO).