Microwave based reversible unfolding and refolding of alcohol oxidase protein probed by fluorescence and circular dichroism spectroscopy ()
1. INTRODUCTION
Microwave radiation (MW) is a non-ionizing electromagnetic radiation growingly used for investigating the properties of biomaterials due to its localized and rapid action of transmitting the energy in aqueous environment [1]. The MW energy is transmitted in the form of heat, while the parallel concept of athermal effect of MW is a subject of much discussion [2-7]. One of the possible explanations for the athermal effects of MW irradiation is the direct energy transfer from the electromagnetic field to the vibrational modes of macromolecules altering their possible conformation [8]. The MW has only few reports on its effect on protein conformation and most of these studies focus on the irreversible effects on proteins [9-12]. Moreover, the MW-based denaturation studies are carried out with small proteins [1,7,9,11]; while the similar studies on large proteins of multimeric nature is not known. We report here the effect of MW radiation on the structure of a homo octameric Alcohol Oxidase (AO-x). The AOx (alcohol: oxygen oxidoreductases, EC 1.1.3.13) from Pichia pastoris is a massive protein with molecular weight of ~600 KDa, each subunits consisting of a noncovalently bound flavin adenine dinucleotide as a cofactor [13]. The subunit molecular mass of AOx has been reported as ~75 KDa and association of the subunits attributed to the hydrophobic interactions [14].
AOx belongs to the family of glucose-methanol-choline (GMC) oxidoreductases. The structure of AOx contains three major domains, namely an FAD-binding domain, a flavin attachment loop and a substrate binding domain, as well as two minor domains, namely an FAD-covering loop and an extended FAD-binding domain. AOx also contains two specific inserted regions in substrate binding domain that are possibly involved in AOx octamer formation [15]. The FAD-binding domain comprises four sequence regions distributed over the whole primary sequence. One of the regions is a common ADP-binding motif (βαβ) present in most FAD-binding proteins containing the characteristic nucleotide-binding site GXGXXG [13]. Since no three dimensional structure of AOx is available to date, its structure is remain speculative. A model of AOx based on the structure of glucose oxidase from Aspergillus niger was generated, and the structure suggests that the FAD molecule is buried in the protein matrix [16]. Based on the deduced amino acid sequences, the AOx from Pichia pastoris contains 8 Trp and 33 Tyr residues [17]. Secondary structure predictions of AOx suggest that the C-terminal part of the molecule, from residues 311 - 664, has the folding of an eightfold beta/alpha-barrel (TIM barrel) [18].
Folding pathway of small proteins usually follow two state mechanism, N↔U [19,20]. The large proteins composed of multiple domains often refold inefficiently because of the formation of partially folded intermediates that tends to aggregate due to hydrophobic forces and inter-chain hydrogen bonding. The multi domain unfolded large protein undergoes refolding through the formation of intermediate species [21] and leads to the folded state. During refolding of a multi-domain protein, if the inter domain distance is high, there will be weak inter-domain interactions and the domains can fold independently. However if the inter domain distance is smaller, then there will be existence of strong inter-domain interactions, which leads to high probability for aggregation [22].
The protein denaturation in in-vitro was generally performed by the addition of high molar concentrations of Guanidinium hydrochloride (Gdn Hcl) or urea to the protein sample, which causes the destabilization of protein by nullifying the electrostatic or hydrophobic interactions through the formation of hydrogen bonds to the peptide groups [23,24]. The reactivation (refolding) of proteins was occurred by dilution of denaturant with appropriate amount of desired buffer or removal of denaturant by dialysis [25,26]. In the present study we established that the MW based partially denatured AOx protein could be revert back to its native conformation with functionally active state by applying a simple incubation technique.
2. MATERIALS AND METHODS
2.1. Materials
Alcohol Oxidase (AOx) (from Pichia pastoris, 32 U∙mg─1 protein), peroxidase from horseradish (HRP) (1080 U·mg─1 solid) and ABTS (2, 2’-Azino-bis (3-ethylbenzothiazoline-6-sulfonic acid) di ammonium salt) were bought from sigma Aldrich (USA). (±)-10-camphorsulfonicacid (CSA) was supplied along with the CD instrument. Hydrogen peroxide (H2O2), potassium-dihydrogen phosphate (K2HPO4), di potassium hydrogen phosphate (K2HPO4) and methanol were obtained from Merck and all other chemicals were of analytical grade. Stock solution of AOx (1.4 mM) was freshly prepared in potassium phosphate buffer (KPB) and that of peroxidase (POD) 250 U·mL–1 in 0.1 M phosphate buffer pH 7.0 and stored at 4˚C.
2.2. Enzyme Assay
AOx activity was determined by spectrophotometric measurement of the production of H2O2 using a coupled peroxidase reaction with ABTS (ε405 = 36.8 mM–1 cm–1) [27], as chromogen. The 1 ml reaction mixture at 25˚C contained a final concentrations of 96 mM KPB, 2 mM ABTS, 0.00001% (W/W) H202, 0.033% (v/v) methanol, 2.5 units peroxidase and 0.01unit AOx (stock solution 0.1 U∙mL–1 in KPB) as described in Sigma-Aldrich protocol. One unit (IU) of enzyme was defined as the amount of enzyme activity producing 1 µmol of H2O2 per min under the assay conditions.
2.3. Microwave Treatment of AOx
The MW radiation was provided from a standard magnetron of a microwave oven (LG MC-8089 TLR, LG Electronics India) operating at 2.45 GHz and 0 - 900 W, and 1 s - 100 min. The sample holders containing the AOx (0.1 mg∙mL–1) was inserted into the MW oven and irradiated for different time intervals at 900 W and 2.45 GHz. After radiation treatment, the enzyme samples were quickly studied for residual enzyme activities, tryptophan (Trp) fluorescence intensities at 295 nm excitation and Far-UV circular dichroism spectra.
2.4. Fluorescence Measurements
All fluorescence measurements (Perkin Elmer LS55) were carried out at room temperature (RT), 25˚C. The slit width of 5 nm was used on excitation and emission monochromators. Emission spectra were recorded by selective excitation of Trp at 295 nm [28], with an integration time of 1 s, a step size of 0.5 nm, scan range from 300 to 430 nm and each spectrum was the average of four scans. The concentration of enzyme used was 0.1 mg·mL–1and background fluorescence of aqueous buffer was subtracted from sample spectra.
2.5. CD Measurements
The CD spectra of the enzyme were recorded in a Jasco J-815 spectropolarimeter calibrated with (±)–10- camphorsulfonic acid for optical rotation. AOx of 0.1 mg∙ml─1 dissolved in 10 mM KPB was used for the analyses. The spectra were determined from 240 - 190 nm, in 0.01 cm path length suprasil quartz cuvette, at a scan rate of 100 nm·min–1, 1 nm intervals, a time constant of 1 s, and average of 3 - 4 scans. The temperature of the cell in the range –15˚C to 90˚C was controlled by using a peltier temperature control unit. The baseline spectrum was subtracted from the sample and the resultant spectra were smoothed by Savitsky-Golay filter using Jasco spectral analysis. To estimate the content of secondary structure, the spectra were analyzed by Jasco SSE-protein secondary structure estimation program supplied with the instrument [29].
2.6. Gel Filtration Experiments
All gel filtration experiments were carried out at RT on a Sephacryl S-300 (10/50) (Mr cutoff 500 - 1500 KDa globular protein) column using an ACTA FPLC device (GE Healthcare). The column was equilibrated with 3 bed volumes of 50 mM phosphate buffer (pH 7.5). For denaturation experiments the AOx samples (0.1 mg/ml) treated with MW for 10 s, 30 s, 40 s and 60 s were applied to the column and protein elution pattern was observed by monitoring the absorbance at 280 nm.
2.7. Optimization of Refolding Conditions
The MW treated protein sample was incubated at 4˚C under static condition for 24 h to bring back the distorted conformation of the AOx obtained by the MW treatment for different time period to the native conformation. Recovery of AOx enzyme activity was measured as follows. The defined volume of irradiated enzyme solution at different exposition time was incubated by following the above incubation conditions and the activity of enzyme was measured for every 10 min of time interval using ABTS method as described above. The refolding experiments analyzed with spectroscopic measurements were done by following the above incubation conditions and monitored for changes in band position at 222 nm and 215 nm for CD spectra and emission maximum wavelength, intrinsic fluorescence emission intensity of tryptophan in the range from 300 - 440 nm.
3. RESULTS AND DISCUSSION
The activity of AOx was sharply declined with the increasing MW treatment time, and at 30 s of treatment nearly 15% original activity was retained (Figure 1). The rate of decrease in AOx activity with MW treatment time was linear till the 30 s of treatment and was calculated as 13.28 U·s─1 (inset of Figure 1). The observed inactivation of AOx by microwave exposure suggests that the structure of AOx was directly affected by the electromagnetic field. The initial burst phase of inactivation was parallel to the increase in Trp fluorescence emission of AOx (Figure 2), which indicates growing exposure of the Trp residues from the buried protein matrix due to unfolding protein with the increasing the MW treatment time. The rate of increase of Trp fluorescence with increasing the treatment time was 10.61au·s─1 (inset of Figure 2). A red shift of 0.5 nm, 1.5 nm, 2.5 nm, 3.5 nm and 5 nm after excitation of the protein at 295 nm for the respective 10 s, 20 s, 30 s, 40 s and 60 s of MW treatments indicates the progressive unfolding of the AOx protein.
The CD spectra of the native AOx in the far UV-region showed a deep negative double maximum at 208 and 222 nm and a positive peak at ~192 nm indicating the presence of α-helical conformation in AOx (Figure 3). The negative band of comparable magnitude at 215 - 220 nm was the characteristic feature of β-sheets [30], suggesting the prominent β-sheets content in AOx. The estimated secondary structure content of AOx was similar to that of previous study which showed 32% α-helix and 41% β-sheet [16]. Since α-helical and β-sheet fractions were found to account for ~74% of the secondary structuresthe native AOx can be considered as a protein with highly ordered secondary structure and stable conformation [16]. A significant decrease in β-sheet with increasing MW treatment time on AOx protein was observed. Nearly 6% and 41% of the β-sheets of the native enzyme were disappeared after the MW treatment for 10 s and 60 s, respectively. However, there was a significant increase in α-helix content with increasing MW treatment till 40s of treatment time. Nearly 13% and 38% of the α-helix content of the native protein increased after the MW treatment for 10 s and 40 s, respectively. Whereas, at 60 s of
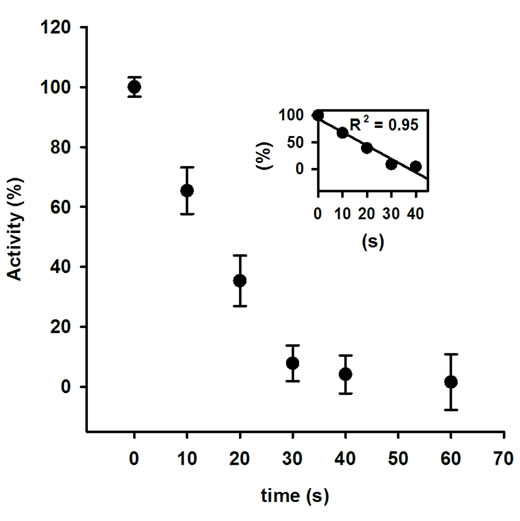
Figure 1. Activity of AOx after exposing to 2.45 GHz microwave irradiation as a function of time.
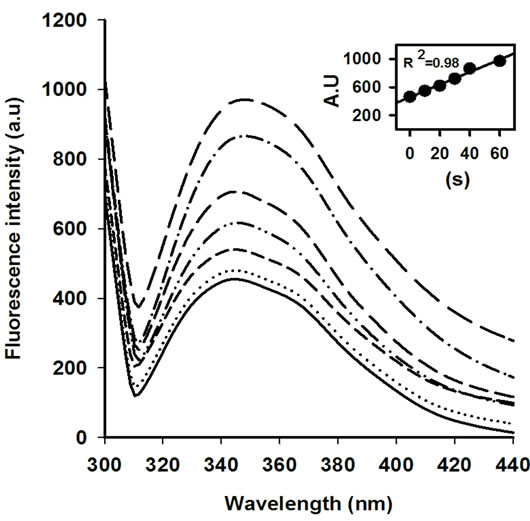
Figure 2. Tryptophan (Trp) fluorescence emission spectra of AOx at 10 s (− − −), 20 s (−∙∙−∙∙−), 30 s (―――), 40 s (−∙−∙−) and 60 s (— — —) of MW treatment in comparison with control (———) excited at 295 nm. The dotted line (∙∙∙∙∙∙∙∙∙) correspond to the refolded AOx obtained by incubating the 10 s MW treated AOx at 4˚C for 24 h.

Figure 3. The Far-UV CD spectra of irradiated AOx at 10 s (−−−−), 20 s (−∙−∙−∙−), 30 s (– – – –), 40 s (−∙∙−∙∙−) and 60 s (— — —) in comparison with (———) control. The dotted (∙∙∙∙∙∙∙) spectrum indicates the refolded AOx obtained by incubating the 10 s irradiated AOx at 4˚C for 24 h.
MW treatment the α-helical structures were decreased by nearly 30% and the turns were completely disappeared with the concomitant increase in the random structures. The results indicate that the intrinsic structure of the protein is extensively damaged by the microwaves energy beyond 40 s of the treatment time.
Refolding experiments were performed to answer the question whether AOx protein irradiated with low frequency MW could be revert back to its native structure. From activity experiment’s as shown in Figure 4, it was observed that an activity recovery of 50% ± 7%, 75% ± 2% and 95% ± 5% were obtained by incubating the 10 s MW treated protein at 4˚C for 15, 25 and 45 min, respectively.
The CD spectroscopic data showed that the distorted secondary structure of AOx generated by the MW exposure for 10 s reverts back closer to the native conformation when the treated sample was incubated at 4˚C for 60 min (Table 1). The renaturation under similar incubating condition for the samples initially treated with MW for 20 and 30 s was partial whereas, it was void for the samples treated for more than 40 s as confirmed by CD analyses.
The results obtained on MW induced conformational transitions of AOx, studied by intrinsic Trp fluorescence and far-UV CD was shown in Figure 5. The unfolding transition monitored by observing the variation of ellipticity at 222 nm and intrinsic tryptophan fluorescence at around 340 nm against different MW exposure time shows that the unfolding of AOx under MW irradiation

Figure 4. The percentage of AOx activity recovered in comparison with control (─■─) after incubating the MW treated enzyme samples at 4˚C for different time intervals. The numbers on the curves indicates the time of exposure to MW in sec.

Figure 5. Unfolding transition curves of AOx as a function of MW radiation exposure time by following fluorescence λmax, and m˚ ellipticity at 222 nm.
occurs without forming any intermediate protein structure. It is known that the MW treatment induces the conformational changes by stimulating the coherent intrinsic dynamics in globular protein molecules [7]. The MW radiation effect observed here is probably mediated by the water molecules attached to the protein surface. The protein bound water absorbs MW radiation in the range of 1 - 15 GHz [31,32] as compared to the free water molecules that absorb at 19 GHz [1]. The MW of 2.45 GHz used in this investigation is thus fall in the range of bound water absorption band. The MW energy received by the protein bound water molecules perturbed the pep-

Table 1. The secondary structural contents of AOx obtained by MW treatment carried out for different time intervals.
tide bond energy of the AOx protein that triggered the rapid conformational change to the protein molecules that skip any intermediate structural transition state of the protein.
The subunits of AOx protein did not detached during MW irradiation. To confirm the fact we performed sizeexclusion chromatography of AOx protein treated with MW for different time period. As shown in Figure 6, the

Figure 6. AOx unfolding induced by MW treatment monitored by gel filtration chromatography. Native AOx with a final concentration of 0.1 mg/ml in the phosphate buffer was treated with MW for different time period (0 s, 10 s, 30 s, 40 s and 60 s) and the protein was applied on to Sephacryl S 300 column equilibrated with 50 mM phosphate buffer. The elution profile was monitored in time scale at 280 nm. The lines of Green, Pink, Black, Red, and Brown colore correspond to 0 s, 10 s, 30 s, 40 s and 60 s of MW treatment time, respecttively and the numbers on the peaks correspond to the elution time of the protein in min.
protein treated for 0 s, 10 s and 30 s were eluted as single peak with corresponding retention time of within 21.2 - 22.0 min. The protein treated for 40 s generated two distinct peaks with retention times of 17.1 min and 21.4 min. The decrease in retention time implies that a fraction of the native protein lost its globular structure due to distorted secondary structure caused by MW and possible partial aggregation of the structurally distorted proteins. The protein obtained by the increased MW treatment time of 60 s eluted as a single peak with retention time of 17.1 min, indicating complete denaturation and possible associated aggregation of the protein. The increasing unfolding of AOx protein with the prolong MW treatment exposes hydrophobic residues of the protein that may facilitate intermolecular interaction and aggregation of the proteins. The findings confirmed that the MW treatment did not dissociate the AOx protein to stable subunits as evident from the void of any peaks with retention time longer than that of the native protein.
Kinetic measurements were carried out to examine the catalytic properties of the enzyme after different MW treatment times using native sample as reference (Table 2). The enzyme solutions exposed to 10 s MW treatment showed numerical values vary from the native enzyme, while larger exposition time (20 s and 30 s) showed a considerable deviation in the kinetic parameter (Km, Vmax and Kcat). The denatured protein obtained by 10s MW treatment was refolded following the techniques described previously. The kinetic parameters of the resulting refolded protein were found to be closer to those obtained by using the native enzyme.
4. CONCLUSION
Studies on protein folding and unfolding are known to provide critical information on the structure-function relationships of proteins. This investigation has established that MW mediated unfolding of the protein is a

Table 2. Kinetic activity parameters of native and MW treated AOx.
reversible phenomena and the reversibility is effectual at low MW exposure time on the protein. The unfolding process enacted by the MW radiation occurs rapidly without passing through any intermediate transition state under the time scale units employed in this investigation. The reversible refolding techniques demonstrated through this investigation has clear advantages over the conventional chemical based approach owing to its direct nature that void the separation steps for the protein involved in chemical denaturant based approach. This MW based technique may be explored for different proteins to establish it as general technique for protein unfolding-refolding studies.
5. ACKNOWLEDGEMENTS
The financial Assistance of DBT, Govt. of India was used to carry out the work.