Synthesis and Conformational Studies of Some Metacyclophane Compounds ()
1. Introduction
The development of ammonium ion recognition systems has been of great concern over the years because of the important roles that ammonium ions play in both chemistry and biology [1-3]. For example, Lehn and his coworkers reported that C3 symmetry crown ethers bearing ester groups form very stable complexes with primary ammonium ions [2,3]. The calixarenes which are a special type of metacyclophanes formed by a condensation reaction involving a phenol and formaldehyde show properties similar to those of the crown ethers. They have a chalice—like shape with defined upper and lower rims, and a central annulus (Figure 1). It has also been reported that calix[n]arene (where n stands for the number of aromatic rings) derivatives with C3 and C6 symmetry can selectively bind ammonium ions [4-9]. The wellknown calixarene molecules include calix[4]arene, calix [6]arene and calix[8]arene [10-14].
Calixarene synthesis rapidly gained popularity, as it became clear that they offered a relatively simple synthetic route to a wide range of new molecular receptors with potentially exciting ion-binding properties. Calixarene with oxygen donor atoms turned out to be suitable for selectively binding alkali metal ions. Nitrophenol or azophenol moieties on calix[4]arene esters were found to produce lithium selective optical responses while similar ligands were found to be sensitive to amines [7]. The introduction of new coordinating functionalities into the lower rim of a calixarene affects the binding affinities for metal cations, and it is possible to manipulate the various cavities to obtain desired selectivities. Although there has been an extensive study of calixarenes, it is surprising that reports on the preparation of the lower members of the series, for example, calix[3]arenes have been very limited [10].
The greatest challenge that confronts a researcher in the field of calixarenes is the freezing of the conformational mobility of these compounds. Various methods have been adopted to solve this problem which include introducing bulky substituents into either the lower or upper rim of the calixarene molecule [15-17]. Intramolecular bridging of each benzene ring is also considered to be a potential method for freezing the conformation of calix[6]arenes [18]. These methods of freezing the various calixarene conformers result in very rigid structures which serve as good hosts in molecular recognition studies [19-24] hence the need to look for other possible calixarene-analogous hosts. The objective of this research was to synthesise and perform conformational analysis on the [3.3.3]metacyclophanes which possess similar properties as the calixarenes.
2. Experimental
All melting points are uncorrected. IR (KBr or NaCl):
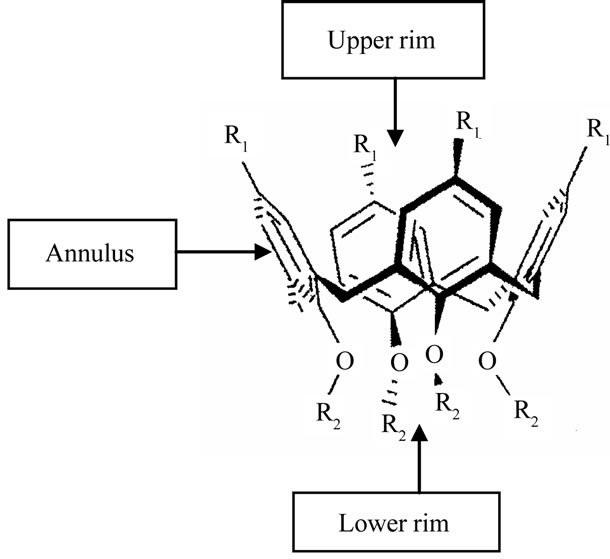
Figure 1. Cone conformation of a typical calix[4]arene.
Nippon Denshi JIR-AQ20M and Shimadzu IR-408. 1H NMR: Nippon Denshi JEOL FT-270 in CDCl3 with tetramethylsilane (TMS) as reference. UV: Hitachi 220A spectrophotometer and Shimadzu UV-240.
MS: Nippon Denshi JMS OISA-2. Elemental analysis: Yanaco MT-5.
2.1. Wolf-Kishner Reduction of 1 (Scheme 1)
A mixture of 1 (320 mg, 0.5 mmol), KOH (200 mg, 3.6 mmol), 100% hydrazine (0.35 ml, 6.2 mmol), and triethylene glycol (5 ml) was heated at 120˚C for 2 h and then at 220˚C for 3 h in a fume chamber. The cooled mixture was poured into water (50 ml), acidified with dilute HCl, and extracted with CH2Cl2 (3 × 50 ml), washed with water (2 × 20 ml), dried with Na2SO4, and concentrated under reduced pressure. The residue was chromatographed on silica gel using benzene as solvent to obtain crude 2 as a colourless solid. Recrystallization from hexane afforded 275.8 mg (0.45 mmol, 90%) of 6,15,24- tri-tert-butyl-9,18,27-trimethoxy[3.3.3]metacyclophane 2 as colourless prisms, m.p. 115˚C - 116˚C. 1H NMR (CDCl3): d = 1.26 (27H, s), 1.78 - 1.85 (6H, m), 2.45 - 2.55 (12H, m), 3.24 (9H, s), 6.95, (6H, s). MS (75 eV): m/z = 612 [M+]. C42H60O3 (612.9): calcd. C 82.31, H 9.87: found C 82.45, H 9.77.
2.2. Demethylation of 2 with BBr3 (Scheme 1)
To a solution of 2 (100 mg, 0.16 mmol) in CH2Cl2 (5 ml) at 0˚C was gradually added a solution of BBr3 (0.2 ml), 2.1 mmol in 1 ml of CH2Cl2. After the reaction mixture had been stirred at room temperature for 4 h, it was poured into ice-water (20 ml), extracted with CH2Cl2 (2 × 10 ml), washed with water (2 × 10 ml), dried with Na2SO4, and concentrated in vacuo to give crude 3 as colourless solid. Recrystallization from benzene afforded 81.3 mg (0.14 mmol, 89%) of 6,15,24-tri-tert-butyl-9,18, 27-trihydroxy[3.3.3]metacyclo-phane 3 as colourless prisms, m.p. 197˚C - 198˚C. IR (KBr): n [cm–1] = 3439 (OH). 1H NMR (CDCl3): d = 1.21 (27H, s), 1.72 (6H, broad s), 2.54 (12H, broad s), 6.85 (6H, s), 7.57, (3H, broad s). MS (75 eV): m/z = 570 [M+]. C39H54O3 (570.9): calcd. C 85.05, H 9.53: found C 82.14, H 9.70.
2.3. Synthesis of Fully “Capped” 4 and Partially “Capped” 5 Products (Scheme 2)
A mixture of 3 (100 mg, 0.18 mmol) and Cesium carbonate (1.14 g, 3.5 mmol) in dry acetone (10 ml) was refluxed for 1 h under N2. Mesitylene tribromide (62.45 mg, 0.18 mmol) was added and the reflux continued for an additional 3 h. After cooling to room temperature, the mixture was filtered. The filtrate was concentrated to give a crude which was chromatographed over silica gel (Kieselgel 60, F254; 100 g) with hexane/chloroform as solvent to afford 15 mg of partially capped product and 96 mg of the fully capped product. Recrystallization from MeOH-CHCl3 (3:1) afforded 8.0 mg (6%) of the partially “capped” product 5 as white solid with m.p. > 300˚C, and 72 mg (60%) of the fully “capped” 4 compound also as a white solid which decomposes at about 256˚C.
2.4. Partially “Capped” Product 5
IR (KBr): n [cm–1] = 2960, 2925, 2850, 1481, 1454, 1260, 1189, 1122. 1H NMR (CDCl3):
d = 1.12 (18H, s), 1.18 (9H, s), 1.74 - 1.98, 2.21 - 2.40, 3.03 - 3.12 (18H, m), 4.40 (2H, d, J = 14.0 Hz), 4.56 (2H, s), 5.35 (2H, d, J = 14.0 Hz), 6.69 (2H, d, J = 1.95 Hz), 6.82 (2H, d, J = 1.95), 6.90 (2H, s), 7.00 (2H, s), 7.18 (1H, s), 8.15 (1H, s). MS (75 eV): m/z = 765 [M+]. C48H61O3Br (765.91): calcd. C 75.27, H 8.03: found C 76.81, H 8.74.
2.5. Fully “Capped” Product 4
IR (KBr): n [cm–1] = 2955, 2863, 1481, 1457, 1363, 1293, 1260, 1190, 1121, 1022, 874. 1H NMR (CDCl3): d = 1.11 - 1.27 (27H, m), 1.72 - 1.99, 2.05 - 2.98, 3.06 - 3.49 (18H, m), 4.34 - 5.30 (6H, m), 6.50 - 7.18 (9H, m). MS (75 eV): m/z = 749 [M+]. C48H61O3·2CH3OH (813.17): calcd. C 80.17, H 8.63: found C 79.34, H 8.27.
Reduction of 6,15,24-Tri-tert-butyl-9,18,27-trimethoxy- [3.3.3]metacyclophane-2,11,20-trione 1 with LiAlH4 (Scheme 3):
To 160 mg (4.216 mmol) of LiAlH4 suspended in 20 ml of dry THF and stood in ice was added drop wise 164 mg (0.250 mmol) of the triketone 1 dissolved in 10 ml of dry THF. After the addition, the reaction mixture was poured into 50 ml ice-water to hydrolyse the excess LiAlH4. The residue was filtered off and the filtrate extracted with CHCl3 (3 × 50 ml), washed with water (2 × 50 ml), dried with Na2SO4, and then evaporated under reduced pressure to obtain 167 mg of residue which was
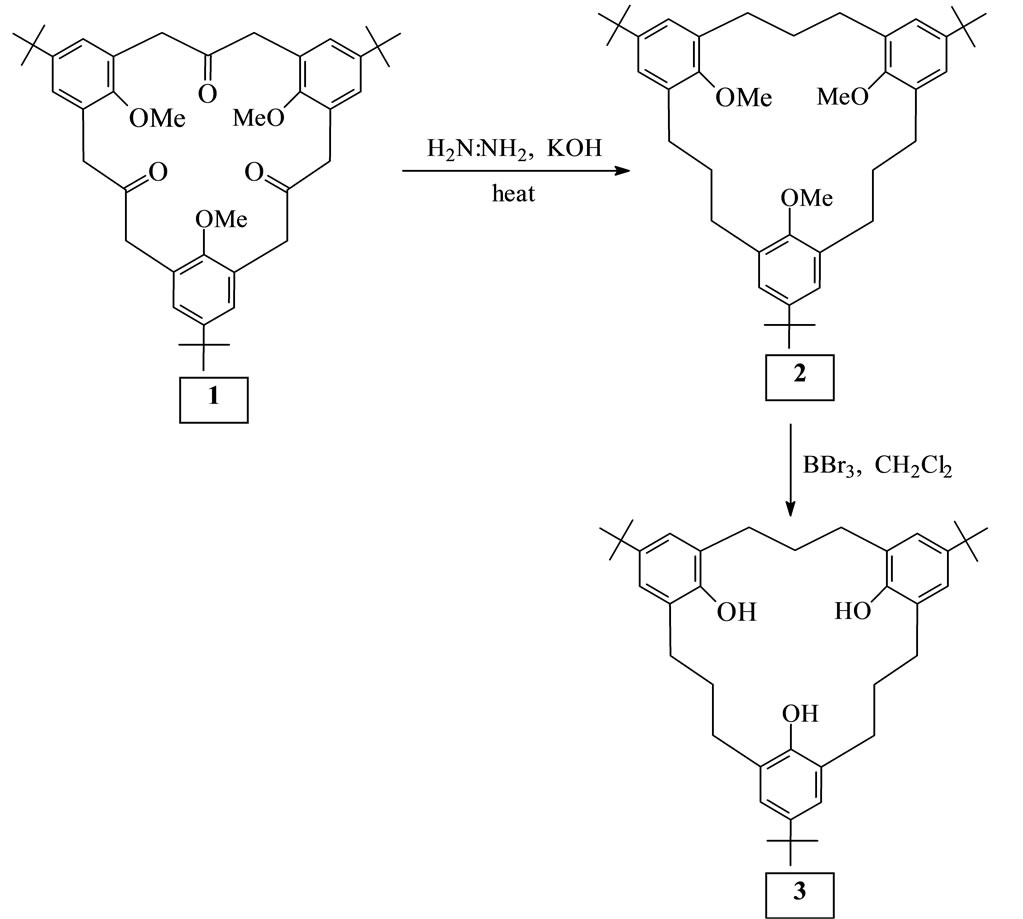
Scheme 1. A Wolf-Kishner reduction of the trione 1 followed by demethylation gave 6,15,24-tri-tert-butyl-9,18,27-trihydroxy [3.3.3]metacyclophane 3.
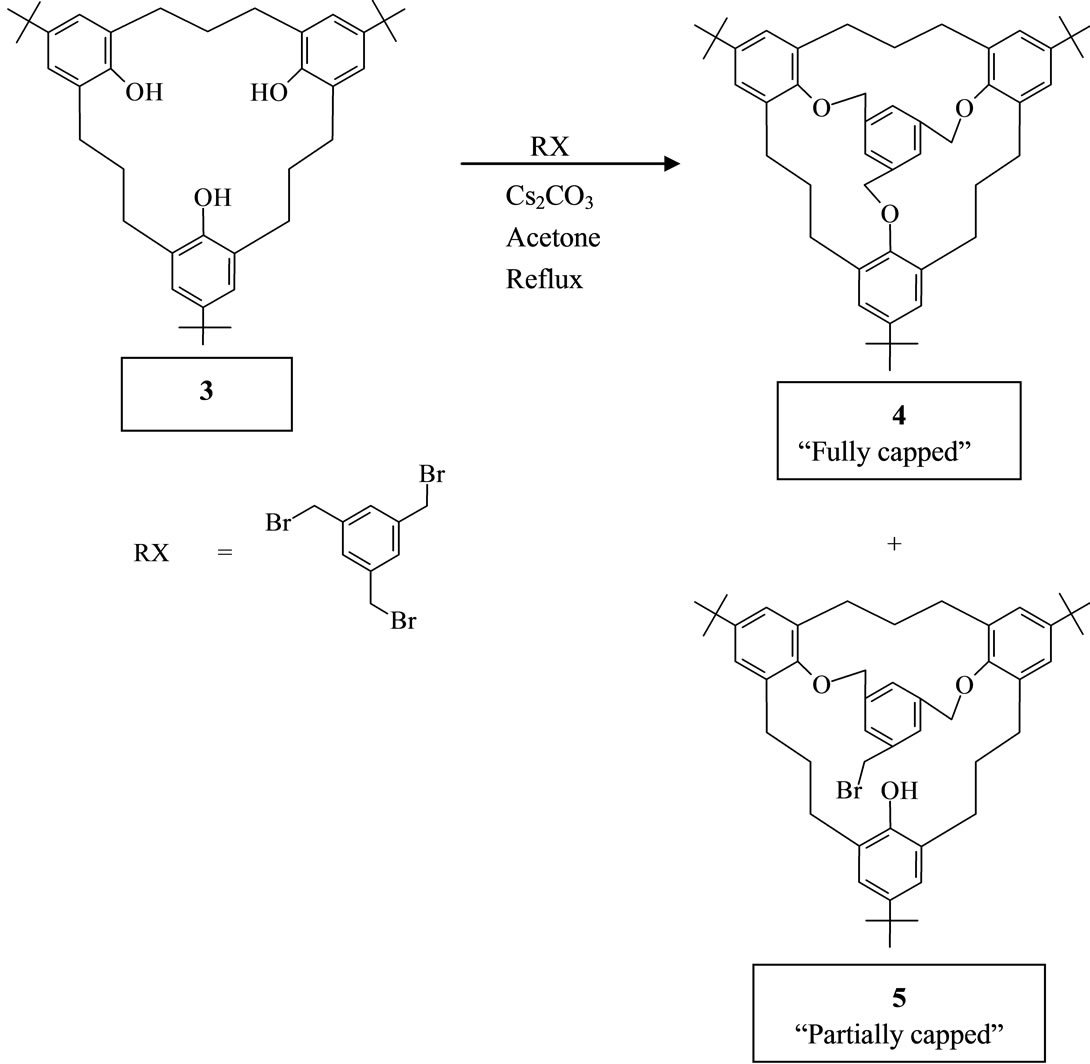
Scheme 2. Bridging the lower rim of metacyclophane 3.
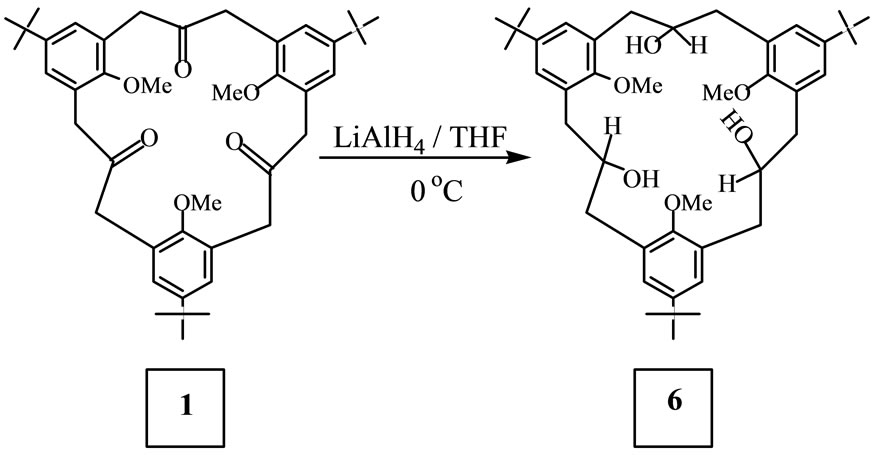
Scheme 3. The reduction of metacyclophane 1.
recrystallized from hexane to obtain 120 mg (0.182 mmol, 67%) of 2,11,20-Trihydroxy-6,15,24-tri-tert-butyl-9,18, 27-trimethoxy[3.3.3] metacyclophane 6 as white crystals with the melting point being 134˚C - 135˚C. IR (KBr): n [cm–1] = 3413 (OH), 2958, 2868, 2826, 1651, 1644, 1603, 1482, 1463, 1432, 1392, 1363, 1307, 1277, 1245, 1207, 1174, 1111, 1045, 1011, 876. 1H NMR (CDCl3): d = 1.28 (9H, s), 1.39 (18H, s), 1.96 - 2.04 (2H, m), 2.24 (3H, s), 2.42 - 2.57 (4H, m), 3.22 - 3.33 (4H, m), 3.75 (6H, s), 4.00 - 4.18 (3H, m), 7.06 (2H, s), 7.10 (2H, d, J = 2.44 Hz), 7.17 (2H, d, J = 2.44 Hz), 7.30 (1H, s, OH), 7.42 (2H, s, OH). MS (75 eV): m/z = 660 [M+]. C42H60O6 (660.93): calcd. C 76.33, H 9.15: found C 76.36, H 9.14.
3. Results and Discussions
Introduction of a trisubstituted benzene unit into the lower rim of 3 via Scheme 2 led to the fully “capped” and partially “capped” compounds 4 and 5.
The partially “capped” trihydroxy[3.3.3.]metacyclophane 5 yielded a rigid product as could be deduced from the 1H NMR spectrum, Figure 2. Thus, a pair of doublets and a singlet representing signals coming from the benzylic protons of the trisubstituted benzene unit showed at δ = 4.50 ppm and δ = 5.40 ppm (for the doublets ), and δ = 4.60 ppm (for the singlet). The -CH2-O protons are the ones responsible for the doublet signals, and the singlet signal could be attributed to the -CH2Br protons [25-27]. The aromatic signals, particularly those coming from the phenolic units confirm the freezing of compound 5 in the “partial-cone” conformation.
In order to study in detail the conformational behaviour of the [3.3.3]metacyclophanes, the triketone 1 was reduced with LiAlH4 yielding the corresponding triol 6, with the OH groups being introduced into the bridge functions (Scheme 3). This triol, which was mobile at room temperature was frozen in the “partial cone” conformation at –50˚C, Figures 3(a) and (b), unlike the parent triketone 1 which remains flexible even at –80˚C. In triol 6, two kinds of methoxy groups were observed with one showing up at a higher field d = 2.24 ppm and the other at d = 3.75 ppm. The former is the methoxy group on the inverted aromatic ring which lies in the shielding zone of the other two benzene rings. In the “partial cone” conformation, the triol 6 may be experiencing weak intramolecular hydrogen bonding between the hydroxyl groups at the bridges and the methoxy groups on adjacent aromatic rings. A further proof for the partial-cone conformation of the triol 6 comes from the aromatic signals as evidenced by the 1H NMR spectra, Figures 3(a) and (b). This situation is somehow similar to that observed in Figure 2.
To further confirm the partial-cone conformation of the triol 6, variable temperature proton NMR studies were carried out on it (Figure 4). It was noticed during these studies that as the NMR temperature was decreased from 27˚C to about –70˚C, changes occurred in certain parts of the NMR spectra. Particularly at δ values 2.0 to 4.0 and 7.0 to 7.5 corresponding to the methylene/methoxy and aromatic signals, it was noticed that these signals which were somehow broad at 27˚C became quite sharp at –50˚C with the aromatic ones showing the characteristic pair of doublets and singlet pattern. These signals appeared at δ values 7.06 (2H, s); 7.10 (2H, d, J = 2.44 Hz) and 7.17 (2H, d, J = 2.44 Hz). The signals for the hydroxyl protons (OH) appeared at δ values 7.30 (1H, s) and 7.42 (2H, s). The signal at δ value 2.40 came from the inverted aromatic ring. The inversion then positioned the OMe group in the shielding zone of the remaining two aromatic rings thus pushing the methoxy signal up-field. These situations gave further proof to the partial cone conformation of the triol 6. The “partial cone” conformation confers on triol 6 a C1 symmetry, which is quite different from the parent triketone which has a C3 symmetry.
4. Conclusion
The studies have revealed that the [3.3.3]metacyclophanes can be easily frozen to show cone and partialcone conformations, a situation similar to that of the calixarenes, for which reason they can serve as “molecular
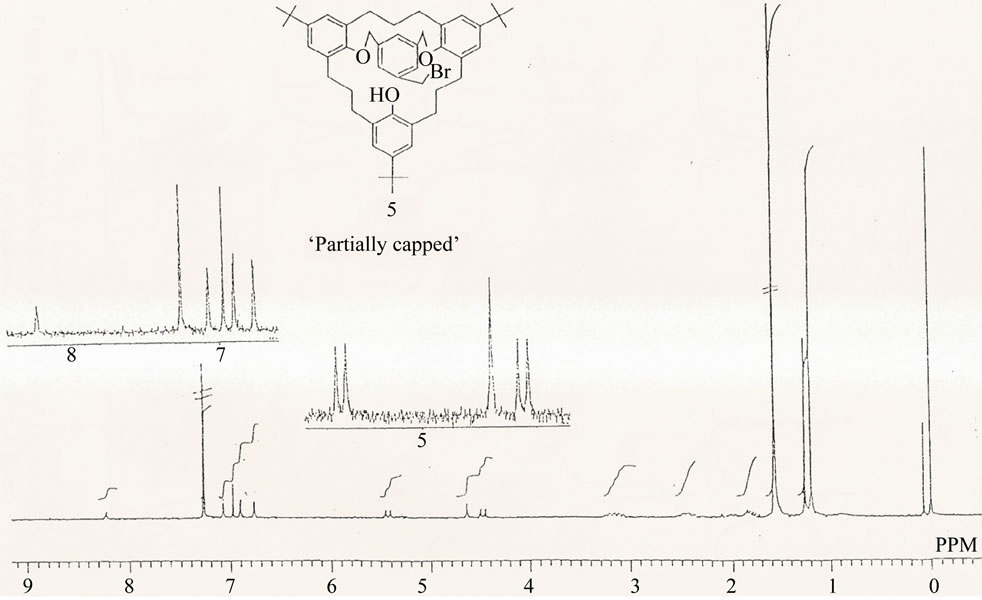
Figure 2. 1H NMR spectrum of the partially capped compound in CDCl3 at 27˚C, 270 MHz, δ (ppm).
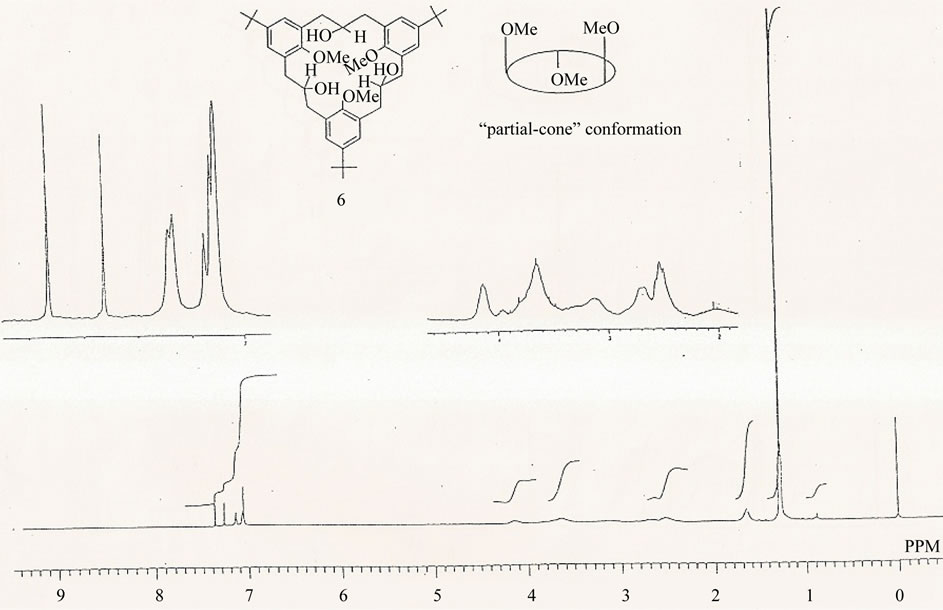 (a)
(a)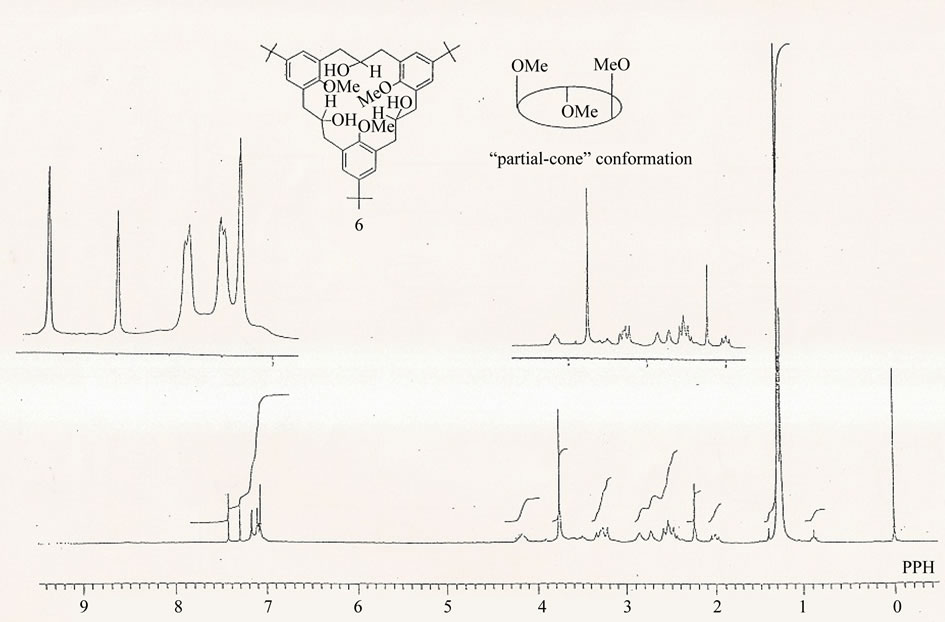 (b)
(b)
Figure 3. (a) 1H NMR spectrum of 2,11,20-trihydroxy-6,15,24-tri-tert-butyl-9,18,27-trimethoxy[3.3.3]metacyclophane 6 in CDCl3 at 27˚C, 270 MHz, δ (ppm); (b) 1H NMR spectrum of 2,11,20-trihydroxy-6,15,24-tri-tert-butyl-9,18,27-trimethoxy [3.3.3]metacyclophane 6 in CDCl3 at –50˚C, 270 MHz, δ (ppm).
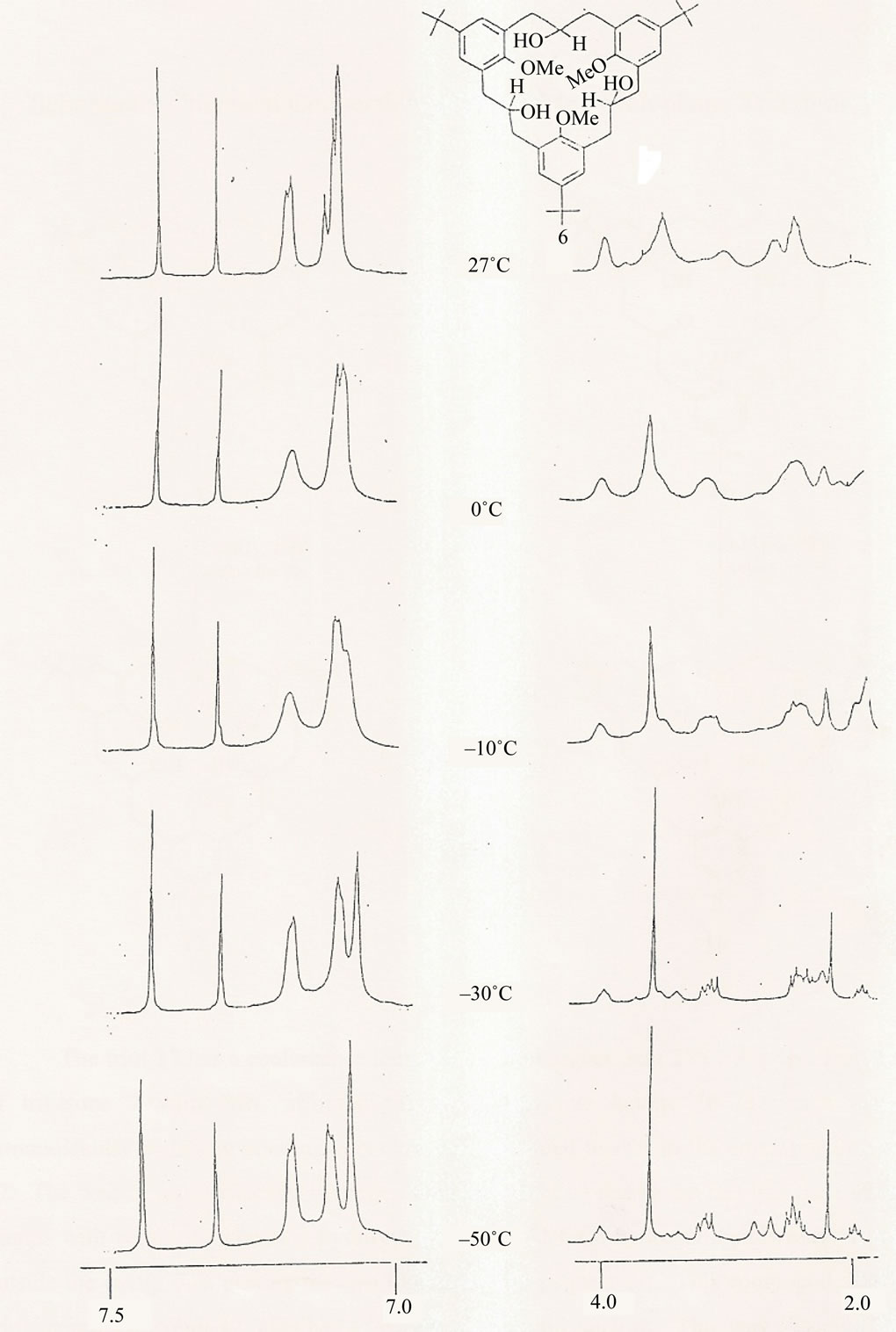
Figure 4. Relevant parts of variable temperature 1H NMR spectrum of 2,11,20-tridroxy-6,15,24-tri-tert-butyl-9,18,27-trimethoxy[3.3.3]metacyc lophane 6 in CDCl3 at 270 MHz, δ (ppm).
baskets” for molecular recognition studies. The stability of the [3.3.3]metacyclophane ring could also be counted as an advantage since it permits the interconversion of functional groups without any ring-opening side reactions.