Common Movement Disorders in Children: Diagnosis, Pathogenesis and Management ()
1. Introduction
The objective of this review is to assist clinicians to be familiar with the clinical presentation, pathogenesis, epidemiology of movement disorders in childhood, to highlight our understanding of the cause and underlying mechanisms and to delineate avenues for future research in this area. The main thrust here is providing tools to reach clinical diagnosis, perform the proper etiological workup and describe available algorithms and treatment strategies.
2. Definition, the Neuroanatomy and the Neurochemical Basis
Most movement disorders can be attributed to dysfunction of the basal ganglia or to regions of the brain that are intimately interconnected with the basal ganglia. The basal ganglia represent a network of interconnected nuclei in the forebrain, diencephalon and midbrain [1,2]. These include the striatum (caudate, putamen, nucleus accumbens), the subthalamic nucleus, the globus pallidus (GP), and the substantia nigra. The striatum and subthalamic nucleus receive the majority of inputs from outside of the basal ganglia. Most of those inputs come from the cerebral cortex, but also form thalamic nuclei. The bulk of the outputs from the basal ganglia arise from the globus pallidus and the substantia nigra pars reticulata. These outputs subsequently inhibit neurons in thalamic nuclei that in turn project to the frontal lobe, and to neurons in the pedunculopontine area.
Most of the movement disorders of basal ganglia origin are circuit disorders that result in impaired ability to facilitate desired movements, inhibit unwanted movements, or both [1,3]. Lesions or developmental abnormalities in one region can result in dysfunction of the entire circuit. Thus, similar clinical syndromes may arise from lesions at different points in the circuit. For example, dystonia may result from lesions in striatum (putamen or caudate), globus pallidus, or thalamus [4,5]. These may subsequently lead to dysfunction in the cerebral cortex and brainstem [6].
Basal ganglia circuitry is not limited to the motor domain, but also includes circuits involved in cognitive and affective function. Thus, many movement disorders of basal ganglia origin are accompanied by cognitive, affective, or behavioral dysfunction.
Most excitatory synapses of the basal ganglia and its connections use glutamate as neurotransmitter. Dopamine is the major neurotransmitter in the nigrostriatal dopamine system. Other important transmitters in these circuits include GABA, acetylcholine, norepinephrine and serotonin [7], and most pharmacologic treatments for movement disorders are targeted at these neurotransmitters.
3. Symptomatology (Table 1)
Movement disorders can be hyperkinetic or hypokinetic. Hyperkinetic movement disorders are characterized by an excessive amount of unwanted or involuntary motor activity. These movements can be either rhythmic such as tremor, or irregular as are tics, chorea or hemiballismus. Stereotypies and myoclonus may be rhythmic or irregular.
Dyskinesia, synonymous to hyperkinetic movement disorders, includes tics, chorea, ballismus, dystonia, myoclonus, stereotypies, and tremor [8]. Tardive dyskinesia, a side effect of dopamine receptor antagonists, can appear even after the drug has been discontinued.
In hypokinetic movement disorders, intentional motor activity is abnormally reduced (bradykinesia) but strength is not significantly diminished while rigidity and postural instability are common. Parkinsonism, is the prototype and the most common hypokinetic movement disorder.
The diagnostic of movement disorder is almost always clinical. Description of the most important phenomena, easily found in textbooks, will not be presented here [7-13].
4. Epidemiology (Table 1)
Most of the movement disorders in children are hyperkinetic. Hypokinetic basal ganglia syndromes are less common and are mainly present in juvenile forms of Parkinson’s disease, Wilson’s disease and Huntington’s disease.
The most common etiology of movement disorders in children is probably cerebral palsy (CP), affecting 2.5 in 1000 live births in the United States [14]. Dystonia or choreo-athetosis are seen in dyskinetic cerebral palsy [15]. Dystonia starting in adolescence may be the presentation of Early-Onset Primary Dystonia (DYT1) [16].
CP is also the most common cause of chorea followed by medications side effects and Sydenham’s chorea (SC). SC is now relatively infrequent in developed countries, attributed in part to aggressive antibiotics treatment against group A streptococci, but still prevalent in the third world [13,17-19].


Table 1. The clinical phenotype of movement disorders and their anatomical correlation.
Tics are very common in children. About 5% of schoolaged children have tics, with an estimated prevalence of 4% - 19% [12]. The estimated prevalence of TS is 3.0 per 1000, three times more in boys than in girls, and approximately twice as common in adolescence than in younger children [20-22]. Tics are usually associated with comor-bidities such as attention deficit hyperactive disorder (ADHD), learning disabilities, pervasive development disorder (PDD) or obsessive compulsive disorder (OCD) [12].
Prevalence of essential tremor in childhood is unknown [14]. Childhood onset is present in up to 15.5% of adult Asian patients with essential tremor [23]. In an epidemicological study in the city of Kolkata, India, there were 6/184 cases within the age group of 10 - 19 years (prevalence of 0.70/1000) [24]. Age of onset is around 8 - 10 years [25]. In a pediatric movement disorder clinic, 19% of patients had tremor as the sole or main feature [14], and up to 50% of individuals diagnosed with essential tremor may report an onset during childhood [26].
Myoclonus in children, mostly of epileptic origin, is beyond the scope of this review.
Drug induced movement disorders are prevalent (Table 2). Besides side effects to a prescribed drug, such as acute dystonia from metoclopramide [27], a movement disorder in a young child is not infrequently due to accidental ingestion of a drug taken by another household member.
It is important to remember that many movement disorders in young children are benign and transient. While present in PDD or Rett syndrome, stereotypies are also frequent in normal children [13].
5. Causes for the Differences between Movement Disorders in Children and Adults
Three factors set the nervous system of the pediatric age group apart from the mature adult brain: the biology of the young brain; environmental factors that are unique to this age group; genetic abnormalities that inherently present either early or late during life.
1) In the newborn brain, a less effective blood brain barrier (BBB), an impaired cerebrovascular autoregulation, and the high metabolic demands of the neonatal period are different than in the adults. Glutamate receptor density and activity are high in the perinatal period, creating a potential for a devastating effect when energy failure occurs. Overstimulation of these receptors can provoke excitotoxicity leading to death of neurons astrocytes or oligodendrocyte. Microglial activation by excitatory amino acids and leukocyte migration initiates an inflammatory response leading to an increase in regional cerebral blood flow and promoting astrocyte and oligodendrocyte injuries [28,29].
2) The non mature brain exposed to hypoxia-ischemia during birth can present later dyskinetic CP. Comparatively, adults who have sustained a severe hypoxia-ischemia, mainly after cardiac arrest, rarely suffer from movement disorders [30]. Another example is pharyngitis due to group A streptococcus that is very frequent in children and uncommon in adults.
3) Genetic background. Specific metabolic abnormalities such as glutaric aciduria type 1, Glucose transporter 1 (GLUT1) and Leigh disease occur exclusively in the young age. Most of the common genetic diseases involving the basal ganglia such as Wilson’s disease, Huntington’s disease or DYT1usually have an adult onset. Others may have a deleterious effect upon the developing brain, but none during adulthood. Thus, in Lesch Nyhan syndrome high level of uric acid causes at one year of age self mutilation, dystonia, choreoatheosis and ballismus but is not associated with any neurological signs in adults [31,32].

Table 2. Drug induced movement disorders.
There are still many conditions that present in childhood and cannot be explained by these three factors or a combination of them. For example, onset of a primary tic disorder in adulthood is rare [10], while tics are very frequent in childhood. Some of the movement disorders are specifically childhood age dependent and transient such as jitteriness, simple tics, spasmus nutans, transient idiopathic dystonia, or benign myoclonus of infancy [33].
6. Pathogenesis and Description of Common Conditions
Basal ganglia of the developing brain are susceptible to several insults, the most prevalent being hypoxia-ischemia, hyperbilirubinemia and infection-associated.
6.1. Hypoxia-Ischemia
Abnormalities within the basal ganglia are observed in up to 84% of patients with perinatal hypoxia-ischemia culminating in shrinkage of the striatum [34]. In the acute phases necrotic or apoptotic neurons can be identified. Later larger zones of necrosis can lead to severe neuronal loss and glial scars. Defects in myelination can result in a marbled gross appearance (hypermyelination-status marmotratus) or in dysmyelination [35].
Vulnerability to hypoxia-ischemia is mainly a feature of the border zones between the major cerebral arteries (“watershed”). In the striatum, neuronal death is probably related to the density of the excitatory glutamate receptors. Striatal GABA-ergic neurons are also sensitive to asphyxia while astrocytes and oligodendrocytes, expressing glutamate receptors as well, participate in basal ganglia injury [34]. Neurons die by glutamate-mediated excitotoxicity involving downstream caspase-dependent and caspase-independent cell death pathways [36]. Delayed apoptosis can extend over days to weeks.
MR-spectroscopy analysis with measures of N-acetylaspartate (NAA) and choline concentrations can identify basal ganglia damage in severely asphyxiated neonates and can help to predict unfavorable outcome [37].
6.2. Hyperbilirubinemia
The basal ganglia of the newborn are very susceptible to increased blood levels of bilirubin. Lesions of the GP, substantia nigra, subthalamic nuclei, as well as cochlear, and oculomotor nuclei, hippocampus, and cerebellum have been demonstrated in kernicterus. Marked loss of neurons, demyelination, and prominent gliosis are present [38,39].
6.3. Infections
The influence of environmental factors unique to the pediatric age group is illustrated by the basal ganglia dysfunction following streptococcal infections. The basal ganglia are the major target of immune-mediated post strep-tococcal infection CNS damage causing SC. Group-A β-haemolytic streptococcal (GABHS) infections (usually pharyngitis) provoke synthesis of antibodies that cross-react between the rheumatogenic strains of streptococci and human basal ganglia epitopes. Such antibodies are present in 50% - 90% of patients with SC [17,40]. The possibility that PANDAS (pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections) is also a post streptococcal disorder in some patients has been raised by some [41]. This is also relevant for encephalitis lethargica, where autoantibodies reactive against basal ganglia antigens are present in 95% of patients, suggesting a post infectious dysimmune mechanism [42].
Infective pathogens such as herpes simplex and measles viruses can rarely involve the basal ganglia as part of acute or chronic encephalitis [43].
6.4. Cerebral Palsy (CP)
CP is a group of disorders attributed to non-progressive disturbances that occurred in the developing fetal or infant brain [44]. CP is generally categorized into spastic, extrapyramidal/dyskinetic, and mixed [15,45]. Dyskinetic CP is more likely to originate closer to pregnancy term or in early infancy. Perinatal asphyxia in term infants accounts for less than 15% of CP in developed countries with a higher incidence in underdeveloped areas [46]. The severity of the MRI and histopathological pathology in the thalamus and basal ganglia correlates with the clinical outcome. A mild abnormal pattern gives a purely dyskinetic CP, while the moderate form results in dyskinetic spastic CP and severe types are associated with spastic CP. On MRI, hyperintense signal and atrophy in the putamen and thalamus is characteristic of asphyxia, while the presence of abnormal signal in the globus pallidus is strongly associated with genetic-metabolic diseases [45]. Diagnosis is dependent on the medical history with evidence for neonatal hypoxia-ischemia [44]. While the majority of children with CP present with symptoms as infants or toddlers, symptom onset may be delayed up to 2 years in dystonic CPs [47]. The clinical picture of dyskinetic CP evolves gradually from diffuse hypotonia with active reflexes in infancy to choreoathetois/dystonia during childhood [34].
6.5. Kernicterus
Kernicterus remains a significant problem, due in part to earlier hospital discharge of neonates and relaxation of rigorous treatment criteria for hyperbilirubinemia [38]. Severe damage can occur with relatively low total serum bilirubin levels in preterms infants [48]. By 2 weeks of age, the infant usually has developed hypertonia, opisthotonus, extensor spasms, and sometimes seizures. By 4 years of age athetosis, dystonia, rigidity, tremor, hearing loss, loss of dental enemal and mental retardation are evident.
6.6. Drugs (Table 2)
Movement disorders in children can be caused by a variety of medications. Of particular importance are the side effects associated with antiepileptic drugs that can cause tremor, tics and chorea [17,49]. Stimulants can produce tics and less commonly chorea [49]. Theophylline can induce tremor and to a lesser degree chorea [17,49].
Dopamine receptor blockers such as typical neuroleptics or atypical antipsychotics can give all types of movement disorders [49]. Hypokinetic syndrome with Parkinsonism, frequent with typical neuroleptics is also present with atypical antipsychotics such as risperidone. Dystonia is seen more with typical neuroleptics, still in use for their low cost [50].
Four studies involving 611 children (mean age 10.2 years) with autism or disruptive behavior disorders, showed that annual tardive dyskinesia rates were 0.35% with second generation antipsychotics [51]. In 783 children receiving risperidone, quetiapine, or olanzapine for a mean of 329.6 days, the annualized tardive dyskinesia rate for risperidone was 0.30%. Tardive dyskinesia resolved within weeks after antipsychotic discontinuation [52].
6.7. SC
This is one of the major criteria of rheumatic fever (RF). Onset is usually around the age of 8 - 9 years, with a female preponderance [17]. It can be bilateral but 20% of patients have hemichorea. Behavioral abnormalities are common and might mask the movement disorder which can be discrete. Diagnosis is based on the clinical context and requires exclusion of other conditions. SC is usually a self limited disorder but recurrences, probably not all related to RF, are described in up to 40% of the patients [19,53].
6.8. PANDAS
Children with PANDAS show dramatic onset of motor or vocal tics, obsessions, and abnormal motor activity with temporal relation to a GABHS infection. [41] The entity of PANDAS remains controversial and it is not clear that it represents a unique subset of the larger spectrum of prepubertal patients with these neuropsychiatric features [54-56].
6.9. Inherited Abnormalities (Table 3)
These are rare causes of movement disorders in children and are beyond the scope of the present review. Most are


Table 3. Some inherited conditions causing movement disorders in childhood that should be considered in the differential diagnosis.
due to already recognized metabolic defects. They are summarized in Table 3 where the typical signs and abnormal laboratory findings are presented in order to rule out these diseases in the differential diagnosis of the more common movement disorders [32,50,57-59].
6.10. Miscellaneous: Tremor, Tics, Wilson’s Disease, Paroxysmal Conditions
Tremor in childhood is seen in a variety of etiologies, mainly metabolic (Table 4) and with drug toxicity (Table 2).
In the immediate postpartum period, excessive tremulous behavior, referred to as jitteriness, involves the entire body. Up to 44% of healthy newborns will exhibit some jitteriness, a benign condition resolving within a few months [26].
The main tremor of childhood is essential tremor. It is characterized by a 4 - 11 Hz frequency that most commonly involves the arms or head, sometimes responsive to ethanol. The neurological examination has to be normal to exclude other processes [26].
Tics (including TS). Most children present with a transient motor tic lasting from 4 weeks to 1 year involving a single muscle in the head, face, neck or arms Tics are more common in boys [12]. Transient tics may also, infrequently, be phonic. Tics may be complex motor (touching, smelling, shaking, jumping) or complex phonic (humming, echolalia, whistling, singing). Coprolalia (uttering obscenities) is reported in 10% of TS patients. If both motor and phonic tics have persisted for more than a year, a diagnosis of TS is appropriate. In TS boys are affected up to 5 times more and have an earlier onset of tics than girls [12].
Wilson’s disease. Although rare, represents the classical treatable movement disorder. Nearly half of all patients present at adolescence or early adulthood with Parkinsonism, tremor, ataxia, titubation, chorea, seizures, dysarthria, and dystonia [58].
Paroxysmal movement disorders are a relatively uncommon but important subset of movement disorders. They include paroxysmal dyskinesias, stereotypies, benign paroxysmal torticollis, spasmus nutans [60]. Sandifer syndrome consists of attacks of gastro-oesophageal reflux with spastic torticollis and dystonic body movements. It is hypothesized that the positioning of the head provides relief from abdominal discomfort caused by acid reflux [61]. Between the attacks the neurological examination is completely normal.
The main and more common possible causes of the various movement disorders are detailed in Table 4.
7. Diagnosis (Tables 1-4)
A meticulous approach is necessary when obtaining the

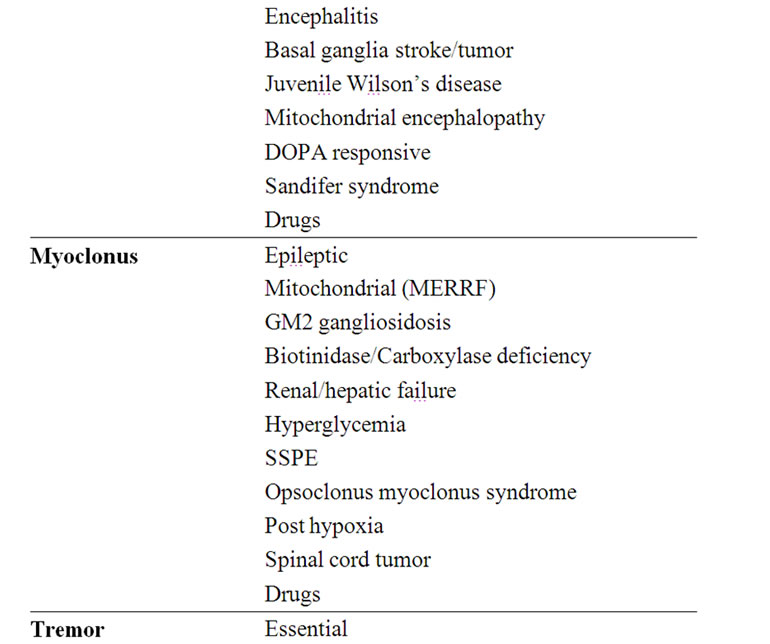

Table 4. Etiology of the various movement disorders.
history: birth and neonatal antecedents such as hypoxiaischemia or kernicterus, positive family history, age at onset, drug history, precipitating factors should be obtained. The most important aspect however is the definition of the clinical phenomenon, with an extended period of observation, using if necessary videotapes. Based on the type of movement disorder and the clinical setup and context, the workup is tailored for each individual patient. Eventually the diagnosis will also rely on clinical hints: e.g.: 1) in movement disorders there is no aura nor post ictal state, but some antiepileptic drugs can improve both epilepsy and movement disorders and the therapeutic response should not be taken as a diagnostic criteria [14]; 2) relation to sleep can help to differentiate the type of movement abnormality (Table 2); 3) tics can be voluntarily suppressed, sometimes for extended periods of time. [11] 4) certain abnormalities may occur in attacks. A meticulous neurological examination is essential in order to classify the various etiologies. While in tics or in benign metabolic conditions (e.g. hyponatremia) there will not be any abnormal sign except for the movement disorder, spasticity can be found in CP, and encephalopathy in diseases such as GLUT1 and glutaric aciduria.
Routine blood tests including biochemistry, thyroid functions, and ceruloplasmin are nearly always needed [9]. When a genetic or ongoing metabolic disease is suspected, specific blood and urine testing, EEG and structural/ functional brain imaging should be performed according to the clinical context. Genetic workup is necessary in suspected cases.
8. Therapy (Table 5)
The literature that summarizes or reviews therapeutic options for movement disorders in children is limited. This is due in part to the lack of controlled therapeutic trials and evidence based data. The therapeutic approach is based on the etiology of the disorder and the phenomenology, and in many instances on both [31,62,63].
8.1. Therapy Based Solely or Mainly on the Etiology
In case of drug toxicity reducing the dosage or avoiding the incriminated compound is the obvious therapeutic approach.


Table 5. Treatment options and doses in each movement disorder.
It is recommended to treat patients with Sydenham’s chorea with penicillin prophylaxis up to the age of 21 years to prevent recurrent attacks, even if they do not have other signs of RF [64]. There are no controlled studies regarding the specific treatment of the chorea. Valproic acid has become the first drug of choice. Carbamazepine can also be effective. Dopamine-receptor blocking drugs are indicated only if other medications fail [17,53,62].
No specific treatment has been proved to be effective in the treatment of PANDAS [41].
The traditional treatment of Wilson’s disease addresses the acute chelation of copper with δ-penicillamine, but more recent less toxic strategies are trientine and zinc or ammonium tetrathiomolybdate [7].
8.2. Therapy Aimed at the Neurological Phenomenology
Dystonia. Dopa-responsive dystonia, an autosomal dominant disorder with worsening of symptoms in the evening dramatically responds to low dose Levodopa. Trihexyphenidyl was not shown to be superior to placebo in a randomized, double-blinded, placebo-controlled, crossover trial conducted with 16 participants with dystonic cerebral palsy. Side effects were common [65].
Essential tremor. Younger children typically have minimal impairment, and the main issue at that age is to reassure the parents. Caffeine consumption should be reduced. Only 61% of 39 children evaluated by Jankovic were medically treated with primidone or propranolol [21].
Tics. Education and reassurance of the parents remains the effective approach. Treatment of tics is needed only when they cause functional impairment. Clonidine and guanfacine are considered as first line medications [66]. Metoclopramide was significantly more effective than placebo in reducing tics in 27 children with TS or a chronic tic disorder [67]. Olanzapine produced statistically significant reductions of aggression and tic severity in ten subjects with a primary diagnosis of TS and a history of aggressive behavior [68]. Both risperidone and clonidine were effective and reduced tics in 5/21 (25%) treated subjects aged 7 to 17 years [69].
One important issue is the treatment of tics appearing or worsening concomitantly with ADHD. Prior recommendations were to avoid methylphenidate in these children. However in a multicenter, randomized, double-blind clinical trial, 136 children with ADHD and a chronic tic disorder were randomly administered clonidine alone, methyphenidate alone, combined treatment, or placebo for 16 weeks. Compared to placebo, therapy was effective: tic severity improved in all treatment groups, mainly with combined treatment. Clonidine was more helpful than methyphenidate but was associated with sedation [70].
8.3. Surgical and Psychological Treatment
Deep brain stimulation (DBS) has been shown to be effective in the treatment of dystonia in adults [71]. Scattered reports of improvement of dystonia with DBS have been documented in children, while patients with dystonia due to perinatal asphyxia might benefit less, due in part to already existing contractures [72].
Severe cases of TS responded to DBS [73,74]. Even neuropsychological and psychiatric co-morbidities may improve after DBS [75].
Botulinum toxin injections can be effective in dystonia, spasticity ans some particularly disabling motor tics [11].
Behavioral therapy has been efficacious in motor stereotypies [13], but not particularly helpful for patients with disabling tics [11].
9. The Future
While the phenomenological arena of movement disorders in the pediatric age group has been mapped quite satisfactory, the field still lacks information and evidence-based data in two aspects. The etiology and pathogenesis of several acquired conditions such as PANDAS and TS is still obscure and should be pursued. More important, double blind controlled clinical studies to evaluate and delineate the best therapeutic approach to most of the conditions described is still missing: what is the best therapy for TS? How should tremor be approached and what is the optimal therapy of tardive dyskinesia in children are examples of questions that should be addressed.
The issue of prevention is, likewise, of importance. Control and prevention of neonatal asphyxia and reducing the permanent damage is a worthy objective for the obstetrician and for this goal the developing of neuroprotective compounds and modalities is of paramount importance. Last, guidelines to avoid the use of agents that may produce movement disorders as transient and permanent side effect should be issued.
NOTES