Spectroscopic Electrochemical Properties and DFT Calculation of 1-Aryltriazenes ()
1. Introduction
1-Aryltriazenes are a class of organic compounds that have garnered significant attention due to their potential as electrochromic materials. Electrochromism refers to the phenomenon where a material’s color changes during an electrochemical reaction, and 1-Aryltriazenes have been shown to exhibit high contrast ratios and various colors in both oxidized and reduced states.
Electrochromic materials change colours depending on the presence or the missing light in response to electrochemical switching [1] . They are in great interest for their potential utility in eyewear protection and data storage devices. Many of the systems used are based on inorganic materials [2] [3] . However, organic materials may overcome many of these issues and as viable alternatives [4] [5] [6] [7] . Among those organic materials, triazene has been considered one of the most important compounds with its standing properties such as optical properties [8] . They have been used to facilitate coupling and functionalized arenes to passivated Si surfaces for applications to semiconductors and nanoelectronics [9] . Triazenes are a unique class of compounds with three consecutive nitrogen atoms in an acyclic arrangement [10] . These compounds can be prepared by diazo copulation between diazonium salt and primary, or secondary amines [11] or Grignard reagents coupled with azide [12] . Due to its eco-friendly, the aqueous synthesis method has been largely used to prepare these compounds [13] . Aryl or alkyl triazenes with alkyl have been studied in medicinal and synthetic chemistry [13] [14] . They are used as iodo-protecting groups [15] [16] [17] [18] [19] in the synthesis of small and macromolecules [1] [20] , protecting groups for amines and diazonium salts [21] [22] , and photoactive substrates [23] . 1-Aryl-3-alkyltriazenes depending on the substituents have been used as precursors for nitrogen-containing heterocycles [24] . Triazenes can exist as a mixture of tautomers. It should be possible to determine the nature, the position of equilibrium provided by the mixture, and the spectrum of the individual tautomer. The tautomers are significantly different [25] . Synthesis of new triazenes derivatives with conductivity and processing properties is an important part of conducting triazene research. This area has attracted great attention due to the improved possibility of the stability, fusibility, and processibility of triazenes without affecting the environmental stability and electrical conductivity. According to their diverse variations of colour and high contrast ratios, functionalized triazenes have become potential electrochromic materials. This latter reversibly changes the colour during the electrochemical reaction, and the phenomenon is called electrochromism [26] . Several conjugated triazenes have colours both in the oxidized and reduced states. Any electrochemical properties studies were done about these 1-aryltriazenes. Spectroscopic studies have been conducted to investigate the electrochemical properties of 1-Aryltriazenes. UV-visible absorption spectra of these compounds have been found to be dependent on the substituents on the aryl group, with the bandgap energy (Eg) defining the onset of the π to π* transition that determines the compound’s color. Electrochemical studies have shown that 1-Aryltriazenes undergo reversible oxidation and reduction processes, with the electrochemical potentials dependent on the substituents on the aryl group. This is the first time that this study was done.
Various solvents have been studied for the solute-solvent interactions occurring in the solvation shell of the solutes have physical and chemical effects on many processes determined by investigating interactions of the solvent with solute, through observation of changes in the electronic absorption spectra obtained [27] . Here, we decided to investigate the effects of solvents on the UV-visible absorption spectrum of triazenes. The biochemical effects of triazenes have been related to oxidative intermediate species [28] . The one-electron oxidative of a triazene gives the primary product, which is expected to be a radical cation according to the reaction (1). This reaction might be performed either by chemical or electrochemical method. Our study aims to contribute to research on the optical properties of triazenes compounds, and the application of quantum chemical calculation methods for the understanding of the molecular structures.
Density functional theory (DFT) calculations have also been used to gain insights into the electronic structure and properties of 1-Aryltriazenes. These calculations have been used to predict the electrochemical potentials and electronic absorption spectra of these compounds, as well as to determine the nature and position of the equilibrium provided by the mixture of tautomers that can exist. The results of these calculations have been found to be in good agreement with experimental data, providing a useful tool for predicting and designing new electrochromic materials based on 1-Aryltriazenes.
Overall, the combination of spectroscopic and computational techniques has provided valuable insights into the electrochemical properties and potential applications of 1-Aryltriazenes as electrochromic materials. Further research in this area may lead to the development of new and improved electrochromic materials with enhanced stability, fusibility, and processibility.
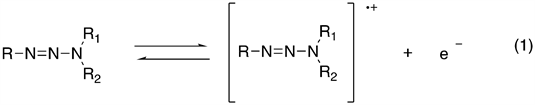
Equation 1. General oxidation reaction of triazenes.
2. Experimental Section
The studied molecules were synthesized and purified according to the common procedure in our laboratory [13] (Figure 1 & Figure 2). The solvents were purchased at Sigma-Aldrich with purity ≥ 98%.
2.1. UV-Visible Spectrophotometer
A UV-Vis spectrophotometer with h quartz cell of 1 cm optical path in the 190 - 850 nm was selected for this study. The custom setting allowed for absorbance measurements for other applications, and a resolution of 0.5 nm was used in this experiment. Three triazenes have been characterized by measuring UV-visible spectra in different solvents such as methanol (MeOH), acetonitrile (ACN), dimethylsulfoxyde (DMSO), and dimethylformamide (DMF). All spectra were determined at 25˚C and corrected for solvent background by calibrating the instrument to the blank solvent.
2.2. Cyclic Voltammetric Study
The electrochemical behavior of the triazenes was investigated using cyclic voltammetry. The experiments were carried out using a Palm Sens potentiostat controlled by PStrace 4 software. The three-electrode mode was employed with Ag/AgCl as the reference electrode and a platinum wire as the counter electrode. The CV measurements were recorded by scanning the potential from 0.5 to +2.0 V at a scan rate of 50 mV s-1 under an N2 atmosphere. The supporting electrolyte used was 0.1 M (Bu4N) ClO4 in 5 mL of acetonitrile (ACN). The use of (Bu4N) ClO4 as the supporting electrolyte was to enhance the electrochemical stability of the triazenes.
2.3. Computational Method
A computational study of (E)-1,3,3-triphenyltriazene, (E)-3,3-diisopropyl-1-phenyltriazene and (E)-(4-chlorophenyl)-3-cyclopentyltriazenewas analysed using DFT and TD-DFT methods to investigate the optical and electronic properties of three different triazene molecules. The study used a hybrid exchange
![]()
Figure 1. Reaction for obtaining 1,3-disubstituted-1,2,3-triazenes.
![]()
Figure 2. Structures of studied compounds.
correlation functional and basis sets containing diffuse functions on the heavy atoms in the system to obtain accurate results for both the ground and excited states [29] . The ground state geometries of the molecules were optimized using the B3LYP exchange correlation functional and the basis set 6 - 31 G(d,p). Vibrational frequency calculations were performed to ensure the stability of the optimized structures and obtain the zero-point corrections of the energy and the Gibbs free energy [30] [31] . The molecular orbitals and electronic transitions were calculated in solvent Acetonitrile using the polarizable continuum model (PCM). Finally, TD-DFT calculations were performed for the first 50 singlet-singlet excited states using the same functional and basis set [29] . All calculations were implemented with the GAUSSIAN 09 package [32] . Overall, the study employed a comprehensive computational approach to investigate the electronic and optical properties of the three triazene molecules. The results could provide useful insights into their potential applications in various fields.
3. Results and Discussion
3.1. Electronic Properties and UV-Vis Absorption
The UV-Visible absorption studies of three triazenes in different solvents such as MeOH, ACN, DMSO and DMF were done as shown in Table 1. The Ionization energy, Electron affinity, Chemical potential, and Energy gap were provided in Table 2.
The electronic transitions of the three compounds were studied using the TD-DFT approach in acetonitrile, and the calculated absorption wavelengths and excitation energies are presented in Table 3. The experimental UV spectra of the compounds were also measured in solvents with varying polarities, and one absorption maximum was observed in the wavelength range of 297 - 350 nm for all the solvents.
The absorption wavelength of a compound can be influenced by the solvent, with an increase in solvent polarity often resulting in a decrease in wavelength for π–π* transitions (Figure 3). In the case of (E)-1,3,3-triphenyltriazene, the intensity of the absorption spectra decreased in the order CAN > DMF > MeOH > DMSO, indicating that solvents with oxygen atoms act as auxochromes, i.e., they act as electron donors. The presence of phenyl groups in the conjugate system
![]()
Table 1. UV/Vis spectral data of triazenes in various solvents.
![]()
Figure 3. Experiment UV-visible spectra of (a) (E)-1,3,3-triphenyltriazene (compound I), (b) (E)-3,3-diisopropyl-1-phenyltriazene (compound II) and (c) (E)-(4-chlorophenyl)-3-cyclopentyltriazene (compound III).
also induced absorbance towards high wavelengths. π–π* electronic transitions at longer wavelengths were observed due to the presence of multiple bonds in the benzene ring and the triazene unit.
The calculated maximum absorption wavelengths correspond to the electronic transition between 300 nm and 400 nm from the HOMO to LUMO. The UV-visible spectrum of (E)-1,3,3-triphenyltriazene in acetonitrile showed two absorption maxima and three shoulders in the range of 150 to 400 nm, with the most intense absorption corresponding to the HOMO → LUMO transition at a wavelength of 351.28 nm (3.5295 eV). This value agrees well with the experimental result of 355 nm.
For the neutral molecules of 3,3-diisopropyl-1-phenyltriazene and 1-(4-chlorophenyl)-3-cyclopentyltriazene in acetonitrile, the UV-visible spectra corresponded respectively to HOMO → LUMO transitions with a large maximum absorption at 299.74 nm (4.1364 eV) and 299.57 nm (4.1387 eV) and the most intense oscillator strength (f = 0.6988) and (f = 0.7372) (Table 2). These results suggest that the electronic transitions of the compounds are highly influenced by the nature of the substituents on the triazene unit, as well as the solvent used in the experiment.
These values can provide additional information about the electronic properties of the compounds, such as their tendency to donate or accept electrons (reflected by the ionization energy and electron affinity), and their overall energy levels (reflected by the chemical potential). The energy gap (Eg) represents the minimum energy required to promote an electron from the valence band to the conduction band and is related to the absorption properties of the compound. A smaller energy gap generally corresponds to a compound that is more likely to absorb light at lower energies (longer wavelengths) and thus appear more colored.
Observing spin degeneration in cationic molecules indicates that a magnetic field is responsible for this effect. It is hypothesized that a local electric field creates this magnetic field, leading to the Zeemann effect. In the oxidized state of the three compounds, we observe the splitting of the HOMO and LUMO orbitals, resulting in different gaps between spins (α and β) (Figure 4). Interestingly, only the oxidized states of these triazenes exhibit color changes. Thus, all elec
![]()
Table 2. Ionization energy, Electron affinity, Chemical potential, and Energy gap.
![]()
Figure 4. Theoretical diagram of energies levels of spin degeneration of compounds (I, II, III) (neutral molecule (E)-1,3,3-triphenyltriazene, (E)-3,3-diisopropyl-1-phenyltriazene, (E)-(4-chlorophenyl)-3-cyclopentyltriazene) and their cationic species molecules.
troactive conductive compounds are potentially electrochromic. The cation form of these compounds has a lowered bandgap, which results in more light absorption in the visible range.
3.1.1. Prediction of the Electrochromic Effect
The DFT calculations provided optimised geometries and UV-Visible spectra, whereas TD-DFT allowed for the simulation of the optical absorption spectra in acetonitrile solvent. The simulated theoretical absorption spectra of neutrals and oxidized molecules are shown in Figures 5-7. The electronic transitions calculated in TD-DFT of all compounds are gathered getting detail in Table 3. The intense transitions colorless—blue in the chromophore of [1,3,3-triphenyltriaz-1-ene] + Figure 5. and colorless—purple in chromophores of [3,3-diisopropyl-1-phenyltriaz-1-ene]+ and [1-(4-chlorophenyl)-3-cyclopentyltriaz-1-ene]+ are good agreement with the electrochromic phenomenon. The different values of the calculated dipole moments between the neutrals and oxidized forms could be used to quantify the charge transfer.
The oxidized and reduced states of these triazenes exhibit distinct colors, and all electroactive conducting compounds have the potential to be electrochromic. Mesomeric effects are observed in the optimized states of the three molecules, showing electronic distributions between their neutral and oxidized forms. In 3,3-diisopropyl-1-phenyltriaz-1-ene and 1-(4-chlorophenyl)-3-cyclopentyltriaz-
![]()
Figure 5. Simulated theoretical absorption spectra of neutrals and oxidized1,3, 3-triphenyltriaz-1-ene.
![]()
Figure 6. Simulated theoretical absorption spectra of neutrals and oxidized3,3-diisopropyl-1-phenyltriaz-1-ene.
![]()
Figure 7. Simulated theoretical absorption spectra of neutrals and oxidized 1-(4-chlorophenyl)-3-cyclopentyltriaz-1-ene.
![]()
Table 3. Geometrical structure of the three optimized compounds (I, II, III) in different levels of oxidation with their energies gap and the dipole moments.
1-ene, the double bond between the two nitrogen atoms near the phenyl group is delocalized, revealing the position of the positive charge after oxidation. However, the mesomeric effect is not observed for the first oxidation of 1,3, 3-triphenyltriaz-1-ene due to the presence of a free doublet on the nitrogen near the two phenyl groups. Additionally, the possible position of cleavage in the molecules can be predicted for all molecules.
Oxydation reaction of cleavage
The R-N=N-N-R, is a symmetrical triazene. The oxidation of this compound can lead to the cleavage of the N-N bond, which results in the formation of two nitrene radicals (R-N·) and molecular nitrogen (N2). This reaction can be represented by the following equation:
R-N=N-N-R + hν → 2R-N· + N2
Equation of oxydation reaction of cleavage.
Where hν represents the energy from light that initiates the oxidation reaction. The two resulting nitrene radicals are highly reactive and can participate in a variety of chemical reactions.
3.1.2. Electron Transfer
In Figure 8, the molecular orbitals responsible for the excited states are presented. The π and π* orbitals are involved in all mentioned transitions. A significant and similar overlap of π bonding orbitals is concentrated on the 1-phenyl groups of the three compounds and one of the two 3-phenyl groups of 1,3,3-triphenyltriaz-1-ene.
The three nitrogen groups are represented by π-type orbitals. The π → π* transition is the first excited state with the highest transition probability, referring to
![]()
Figure 8. Structures of optimized compounds in different levels of oxidation.
their greatest oscillator strength. Overall, these results provide valuable insight into the electronic properties of the compounds and their potential applications.
The projected density of states for 1,3,3-triphenyltriaz-1-ene, 3,3-diisopropyl-1-phenyltriaz-1-ene, and 1-(4-chlorophenyl)-3-cyclopentyltriaz-1-ene. The contribution of the three nitrogen and different groupings (1,3,3-triphenyl, 3,3-diisopropyl-1-phenyl, and 1-(4-chlorophenyl) -3-cyclopentyl) are shown in Figures 9-11. The scale of the isopropyl contribution is 1/4 of the scale shown in
![]()
Figure 9. Projected density of states of 1,3,3-triphenyltriaz-1-ene with the HOMO-LUMO electron transfer representation.
![]()
Figure 10. Projected density of states of 3,3-diisopropyl-1-phenyltriaz-1-ene with the HOMO-LUMO electron transfer representation.
![]()
Figure 11. Projected density of states of 1-(4-chlorophenyl)-3-cyclopentyltriaz-1-ene with the HOMO-LUMO electron transfer representation.
Figure 6. Gaussian distributions with 0.3 eV full width at half maximum were used to convolute the energy levels.
3.2. Cyclicvoltammevoltammetric
The results of a study on the electrochemical properties of three compounds, I, II, and III, using cyclic voltammetry in an electrolyte medium containing 0.1 M acetonitrile (C2H5)4N(BF4). The study found that all three compounds showed a stable oxidation-reversible process with the presence of a pair of redox peaks for each compound.
Compound I showed the presence of a redox couple (0.8 and −0.5 V/Ag/AgCl) linked to the presence of the phenyl group in the compound. Compound II showed the presence of two anodic peaks towards 1.44 and 0.92 V/Ag/AgCl and two reduction peaks around 0.42 and −0.44 V/Ag/AgCl. The values of the potentials of the oxidation peak around 1.44 V/Ag/AgCl and of the reduction of the peak of 0.42 V/Ag/AgCl (Figure 12) are similar to those described in the case of an aliphatic diamine [33] . The redox couple 0.99 and −0.44 V/Ag/AgCl is attributed to the phenyl group. Compound III showed an oxidation and reduction peak respectively −0.27 and −0.8 V/Ag/AgCl attributed to the phenyl molecule (Table 4). This peak shift towards negative potentials could be due to the attractiveness of the chlorine atom.
The study concluded that all three compounds were electroactive and exhibited reversible characteristics with oxidizing/reducing couples. Compounds II and III showed a more positive potential compared to compound I, indicating the establishment of a new type of material with a considerable improvement in redox responses compared to materials in the literature [34] . An additional reduction peak was observed for both complexes at −0.6 V, but the intensity decreased when the scan rate increased, indicating the presence of a chemical reaction.
3.3. Analytical Data of 1,3-Disubstituted 1,2,3-Triazenes
3.3.1. (E)-1,3,3-Triphenyltriaz-1-Ene
The yield of the synthesis was 92%, resulting in a dark brown solid with a melting
![]()
Figure 12. Voltammograms of Compound (I), Compound (II) and Compound (III).
![]()
Table 4. Oxidation and reduction potentials of 1-aryltriazenes.
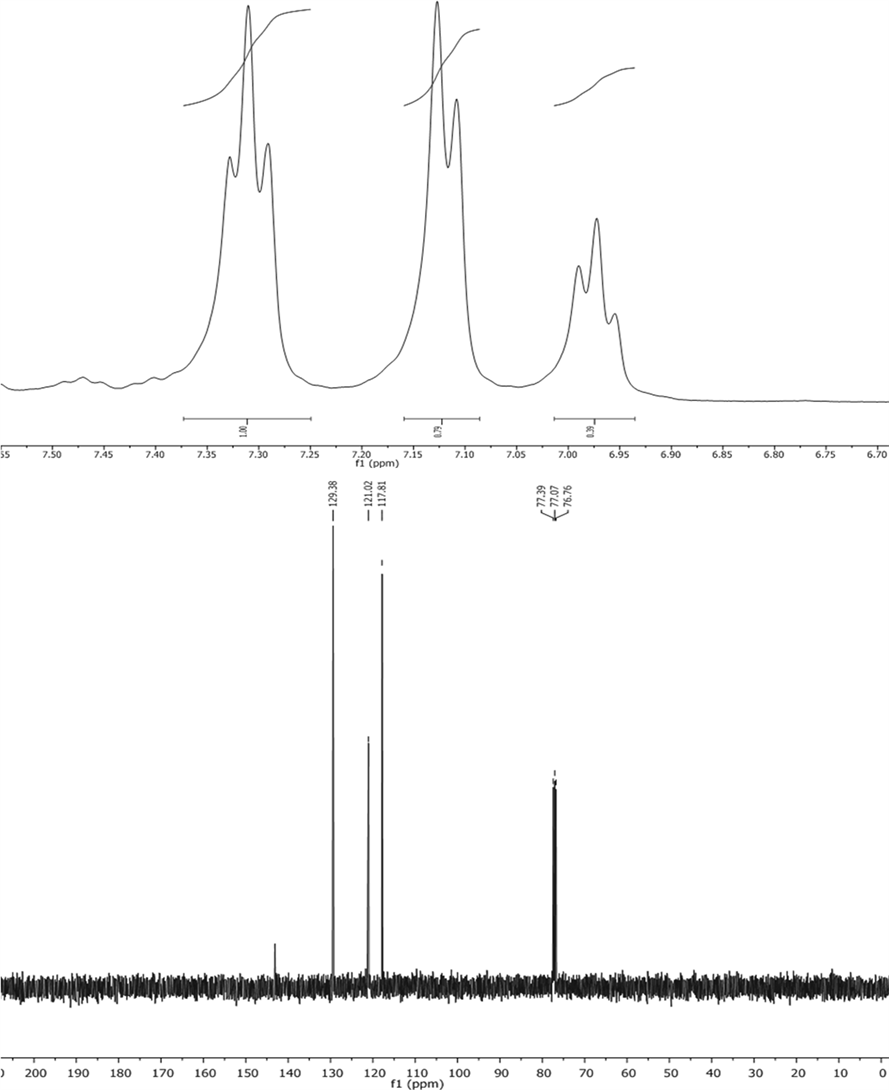
point (Mp) of 98˚C - 104˚C. The molecular formula of the product is C18H15N3 with a molecular weight (MW) of 273.33. Thin-layer chromatography (TLC) showed a retention factor (Rf) of 0.65 using a mixture of AcOEt/Hex in a 1:9 ratio. The proton nuclear magnetic resonance (1H NMR) spectrum of the product, recorded using CDCl3 solvent and a 400 MHz spectrometer, displayed three peaks at chemical shifts (δ) of 7.31 (triplet, J = 7.6 Hz, 3H), 7.12 (doublet, J = 8.0 Hz, 6H), and 6.98 (triplet, J = 7.3 Hz, 6H). The carbon-13 nuclear magnetic resonance (13C NMR) spectrum of the product, recorded using CDCl3 solvent and a 101 MHz spectrometer, displayed five peaks at chemical shifts (δ) of 143.12, 129.39, 121.02, 117.82, and an additional signal at 170.1080, which corresponds to the carbon in the nitrogen-containing ring. The infrared (IR) spectrum of the product, recorded using CDCl3 solvent, displayed peaks at wavenumbers (ν) of 3405, 3381, 3040, 1587, 1491, 1306, 740, and 686 cm−1. The mass spectrometry (MS) analysis of the product, performed using ionization energy (IE+) mode, displayed two peaks at m/z 170.1080 ([C12H11N]+, 100%) and 274.1338 ([M + 1]+, 6%).
3.3.2. (E)-3,3-Diisopropyl-1-Phenyltriazene
The reaction yielded 71%, producing a red solid with a melting point of 40˚C - 45˚C. The compound has a molecular weight of 205.3 and the TLC showed an Rf value of 0.66 using AcOEt/Hex (1:9) as the solvent system. The 1H NMR spectrum (400 MHz, CDCl3) displayed signals at δ 7.45 (2H, d, J = 7.9 Hz), 7.35 (2H, t, J = 7.8 Hz), 7.14 (1H, t, J = 7.8 Hz), and 1.34 (12H, d, J = 7.2 Hz). The 13C NMR spectrum (101 MHz, CDCl3) showed peaks at δ 151.76, 128.77, 124.73, 120.31, 43.12, and 14.5. The IR spectrum (CDCl3) exhibited absorption bands at 2974, 2932, 1403, 1226, 1148, 766, 692, and 538 cm−1. The MS (IE+) showed a molecular ion peak at m/z 206.1660 ([M + 1], 100%).
3.3.3. (E)-(4-Chlorophenyl)-3-Cyclopentyltriazene
Yield: 86%. Yellow solid with a melting point of 68˚C - 70˚C. The molecular formula is C11H14ClN3 and the molecular weight is 223.7. TLC analysis gave an Rf value of 0.2 in a mixture of AcOEt/Hex (1:9). The 1H NMR spectrum (400 MHz, CDCl3) shows signals at 7.12 ppm for two doublets with coupling constants J = 8.7 and 2.2 Hz, 6.61 ppm for two doublets with coupling constants J = 8.8 and 2.1 Hz, 3.51 ppm for a doublet of triplets with coupling constants J = 7.0 and 2.1 Hz, 2.06 ppm for a singlet, and 1.24 ppm for a quartet with coupling constant J = 7.1 Hz. The 13C NMR spectrum (101 MHz, CDCl3) displays signals at 145.06, 129.12, 123.05, 116.28, 65.91, 32.09, and 15.32 ppm. The IR spectrum (CDCl3) shows absorption bands at 2974, 2932, 1403, 1226, 1148, 766, 692, and 538 cm−1. The mass spectrum (IE+) (m/z, %) displays a peak at 206.1660 ([M + 1], 100).
4. Conclusion
In conclusion, triazenes are a unique class of compounds that have various potential applications in different fields, such as medicinal and synthetic chemistry, semiconductors, and nanoelectronics. Organic electrochromic materials, such as triazenes, have attracted significant attention due to their potential use in eyewear protection and data storage devices. With their diverse variations of color and high contrast ratios, functionalized triazenes have become potential electrochromic materials, which change their color during an electrochemical reaction. The effects of solvents on the UV-visible absorption spectrum of triazenes have been investigated to understand the solute-solvent interactions, which play a critical role in many processes. The study also aims to contribute to the research on the optical properties of triazenes and the application of quantum chemical calculation methods to understand the molecular structures. Therefore, further research on triazenes and their potential applications is essential to improve the stability, fusibility, and processibility of these compounds while maintaining environmental stability and electrical conductivity.
Credit authorship Contribution Statement
Insa Seck: Conceptualization, Investigation, Supervision, Writing-original draft. Mohamed Lamine Sall: Investigation, Supervision, Validation and Writing.
Atoumane Ndiaye: Computational calculation, Validation and Writing
Samba Fama Ndoye: Writing
Lalla Aicha Ba: Writing
Matar Seck: Writing
Supporting Information
(E)-1,3,3-triphenyltriaz-1-ene
(E)-3,3-diisopropyl-1-phenyltriazene
![]()
(E)-(4-chlorophenyl)-3-cyclopentyltriazene
![]()
![]()