Environmentally Friendly Syntheses of Imines Applying the Pressure Reduction Technique: Reaction Cases of Less Reactive Amines and Studies by Computational Chemistry ()
1. Introduction
The concept of green chemistry had deeply penetrated into society, and the development of the organic synthetic method that was preferable for green chemistry progressed rapidly in recent years. Syntheses of imine derivatives were no exception and also attempted to be developed with the concept of green chemistry. The imine derivative having a CN double bond in a molecule had received widespread attention in various fields due to its chemical property. Particularly, it was important compound for functional materials, pharmaceuticals, or agricultural chemicals, and there were many reports of its use as an active starting material [1] - [12]. Therefore, the development of the effective synthetic method of the imine derivative was strongly desired in the field of organic synthesis. In response to this request, various methods had been developed. For example, it was well known that a secondary amine could be transformed to the corresponding imine by the oxidizing agent [13] [14]. It was also reported that benzyl alcohol was reacted with an aromatic amine in the presence of the oxidizing agent to give the target product [15]. Although it was similar to this method, the synthetic reaction of the desired imine from benzylamine and an aromatic amine had been reported [16]. Moreover, the synthetic method of a target compound by the rearrangement reaction or the synthesis of the imine derivative by the reaction of nitrobenzene with a corresponding aldehyde had been developed [17] [18]. These methods were considered to be very useful because the desired compounds were obtained in moderate to good yields. On the other hand, the most direct synthetic method of the imine derivative was the reaction of an aldehyde with an amine. This method proceeded with dehydration to give the desired compound [19]. We focused on this reaction, and developed the new synthetic method of the imine derivative under solvent free [20] [21] [22] [23] and catalyst free reaction conditions, previously [24] [25]. In our method, no external energy supply such as heating or microwave irradiation [26] [27] [28] was required, and the target compound was obtained quantitatively using the pressure reduction technique. These facts suggested that our synthetic method was favorable for green chemistry. In this system, when active amines were used as substrates and mixed with aldehydes (1:1, substance ratio) exactly, the reaction proceeded in the short reaction time. However, it was found that the reaction was not completed when this method was carried out under the same reaction conditions using amines having low reactivity. In order to solve this problem, we had studied the effect of using a small excess of amines to aldehydes and the appropriate control of the reaction time. Then, we could find useful and efficient synthetic methods of imine compounds in excellent yields (Scheme).
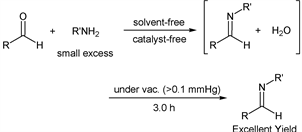
Scheme Solvent- and catalyst-free syntheses of imine derivatives
We will introduce the detail of this study, and also report the result of the investigation of the reaction property by computational chemistry.
2. Experimental
2.1. Chemicals and Instruments
Standard bench top techniques were employed for handling air-sensitive reagent. Liquid aldehydes and all amines were distilled under argon before use. p-Bromobenzaldehyde and p-chlorobenzaldehyde were purified by recrystallization before use. Starting materials were listed in Figure 1. All reactions were carried out under nitrogen atmospheres. All yields of target compounds were isolated yields. ULVAC G-50DA (ULVAC KIKO Inc.) was used for carrying out this reducing pressure operation. IR spectra were recorded on an FT/IR-610 (JASCO) spectrophotometer. 1H-NMR and 13C-NMR spectra were measured on Bruker BioSpin AVANCE III 400 Nanobay spectrometer at 400.1 and 100.6 MHz, respectively. Chemical shifts were given in ppm relative to TMS. Acer Aspire XC-780-N78G (Core i7 CPU, i7-7700@3.60 GHz, 8 GB RAM, 2 TB HDD, OS: Windows 10 (Microsoft Corporation)) was used to run all the calculations. Ab initio calculations (HF/6-31+G*, HF/6-311G*) [29] and DFT calculations (ωB97X-D/6-31G*, EDF2/6-31G*) [30] [31] [32] [33] were carried out by the computer software Spartan ’16 ver. 2.0.7 (Wavefunction Inc.).
2.2. Typical Experimental Procedure 1 (Reaction of p-Chlorobenzaldehyde with n-Propylamine)
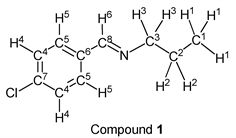
To a stirring p-chlorobenzaldehyde (1.41 g, 10.0 mmol) was added dropwise n-propylamine (0.65 g, 11.0 mmol) at 25˚C. After 3.0 hours, the reaction system was connected to a vacuum pump, the pressure was reduced to >0.1 mmHg (13.3 Pa) and stirred for 3.0 hours to give the desired pure compound (Compound 1) as a clear oil in 98% yield (1.78 g) without any purification. All physical properties of this product were completely consistent with literature values [34] or physical data of the commercially available compound: IR (neat): 504, 820, 1088, 1489, 2837, 2873, 2930, 2961 cm–1; 1H-NMR (CDCl3) δ (ppm): 0.95 (t, J = 7.40 Hz, 3H, H1), 1.72 (sext, J = 7.40 Hz, 2H, H2), 3.57 (t, J = 7.50 Hz, 2H, H3), 7.38 (d, J = 8.44 Hz, 2H, H4), 7.66 (d, J = 8.48 Hz, 2H, H5), 8.23 (s, 1H, H6);
13C-NMR (CDCl3) δ (ppm): 11.87 (C1), 24.02 (C2), 63.50 (C3), 128.85 (C4), 129.22 (C5), 134.80 (C6), 136.36 (C7), 159.47 (C8).
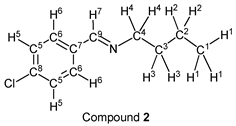
p-Chlorobenzaldehyde (1.41 g, 10.0 mmol) reacted with n-butylamine (0.80 g, 11.0 mmol) to give the corresponding pure compound (Compound 2) in 99% yield (1.93 g) as a clear oil in the same manner of procedure 1. All physical properties of this product were completely consistent with literature values [35] [36] or physical data of the commercially available compound: IR (neat): 505, 820, 1088, 1489, 1647, 2840, 2871, 2930, 2958 cm–1; 1H-NMR (CDCl3) δ (ppm): 0.92 (t, J = 7.40 Hz, 3H, H1), 1.36 (sext, J = 7.40 Hz, 2H, H2), 1.65 (quin, J = 7.28 Hz, 2H, H3), 3.58 (t, J = 7.04 Hz, 2H, H4), 7.35 (d, J = 8.48 Hz, 2H, H5), 7.63 (d, J = 8.48 Hz, 2H, H6), 8.21 (s, 1H, H7); 13C-NMR (CDCl3) δ (ppm): 13.90 (C1), 20.46 (C2), 32.94 (C3), 61.45 (C4), 128.83 (C5), 129.18 (C6), 134.81 (C7), 136.33 (C8), 159.36 (C9).
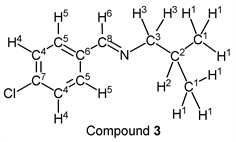
p-Chlorobenzaldehyde (1.41 g, 10.0 mmol) reacted with i-butylamine (0.80 g, 11.0 mmol) to give the corresponding pure compound (Compound 3) in 99% yield (1.93 g) as a clear oil in the same manner of procedure 1. All physical properties of this product were completely consistent with literature values [37] or physical data of the commercially available compound: IR (neat): 506, 827, 1089, 1490, 1647, 2870, 2899, 2926, 2956 cm–1; 1H-NMR (CDCl3) δ (ppm): 0.93 (d, J = 6.68 Hz, 6H, H1), 1.98 (sep, J = 6.68 Hz, 1H, H2), 3.41 (d, J = 6.64 Hz, 2H, H3), 7.36 (d, J = 8.48 Hz, 2H, H4), 7.65 (d, J = 8.48 Hz, 2H, H5), 8.18 (s, 1H, H6); 13C-NMR (CDCl3) δ (ppm): 20.67 (C1), 29.55 (C2), 69.74 (C3), 128.82 (C4), 129.22 (C5), 134.81 (C6), 136.32 (C7), 159.47 (C8).
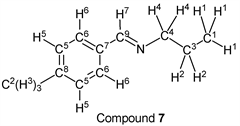
p-Bromobenzaldehyde (1.85 g, 10.0 mmol) reacted with n-propylamine (0.65 g, 11.0 mmol) to give the corresponding pure compound (Compound 4) in 98% yield (2.22 g) as a clear oil in the same manner of procedure 1. All physical properties of this product were completely consistent with literature values [38] or physical data of the commercially available compound: IR (neat): 499, 818, 1011, 1066, 1487, 1589, 1648, 2836, 2930, 2960 cm–1; 1H-NMR (CDCl3) δ (ppm): 0.95 (t, J = 7.40 Hz, 3H, H1), 1.72 (sext, J = 7.32 Hz, 2H, H2), 3.57 (t, J = 6.96 Hz, 2H, H3), 7.54 (d, J = 8.48 Hz, 2H, H4), 7.60 (d, J = 8.52 Hz, 2H, H5), 8.22 (s, 1H, H6); 13C-NMR (CDCl3) δ (ppm): 11.84 (C1), 23.96 (C2), 63.49 (C3), 124.78 (C4), 129.43 (C5), 131.78 (C6), 135.18 (C7), 159.56 (C8).
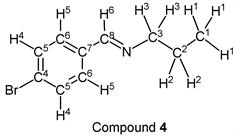
p-Bromobenzaldehyde (1.85 g, 10.0 mmol) reacted with n-butylamine (0.80 g, 11.0 mmol) to give the corresponding pure compound (Compound 5) in 98% yield (2.36 g) as a clear oil in the same manner of procedure 1. All physical properties of this product were completely consistent with literature values [36] : IR (neat): 499, 974, 1011, 1067, 1486, 1647, 2839, 2871, 2930, 2957 cm–1; 1H-NMR (CDCl3) δ (ppm): 0.92 (t, J = 7.36 Hz, 3H, H1), 1.36 (sext, J = 7.40 Hz, 2H, H2), 1.65 (quin, J = 7.24 Hz, 2H, H3), 3.58 (t, J = 7.04 Hz, 2H, H4), 7.51 (d, J = 8.48 Hz, 2H, H5), 7.57 (d, J = 8.52 Hz, 2H, H6), 8.19 (s, 1H, H7); 13C-NMR (CDCl3) δ (ppm): 13.90 (C1), 20.45 (C2), 32.90 (C3), 61.46 (C4), 124.76 (C5), 129.41 (C6), 131.77 (C7), 135.22 (C8), 159.44 (C9).
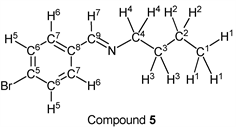
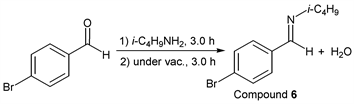
p-Bromobenzaldehyde (1.85 g, 10.0 mmol) reacted with i-butylamine (0.80 g, 11.0 mmol) to give the corresponding pure compound (Compound 6) in 99% yield (2.38 g) as a clear oil in the same manner of procedure 1. All physical properties of this product were completely consistent with literature values [39] : IR (neat): 500, 822, 1011, 1067, 1486, 1590, 1649, 2839, 2870, 2898, 2926, 2955 cm–1; 1H-NMR (CDCl3) δ (ppm): 0.93 (d, J = 6.72 Hz, 6H, H1), 1.98 (sep, J = 6.68 Hz, 1H, H2), 3.40 (d, J = 6.64 Hz, 2H, H3), 7.52 (d, J = 8.48 Hz, 2H, H4), 7.58 (d, J = 8.52 Hz, 2H, H5), 8.16 (s, 1H, H6); 13C-NMR (CDCl3) δ (ppm): 20.69 (C1), 29.55 (C2), 69.77 (C3), 124.78 (C4), 129.48 (C5), 131.79 (C6), 135.25 (C7), 159.57 (C8).
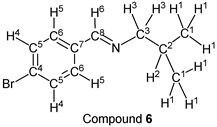
2.3. Typical Experimental Procedure 2 (Reaction of p-Tolualdehyde with n-Propylamine)
To a stirring p-tolualdehyde (1.20 g, 10.0 mmol) was added dropwise n-propylamine (0.65 g, 11.0 mmol) at 25˚C. After 5.0 hours, the reaction system was connected to a vacuum pump, the pressure was reduced to >0.1 mmHg (13.3 Pa) and stirred for 3.0 hours to give the desired pure compound (Compound 7) as as a clear oil in 98% yield (1.58 g) without any purification. All physical properties of this product were completely consistent with literature values [40] or physical data of the commercially available compound: IR (neat): 499, 814, 969, 1648, 2833, 2873, 2928, 2960 cm–1; 1H-NMR (CDCl3) δ (ppm): 0.94 (d, J = 7.40 Hz, 3H, H1), 1.72 (sext, J = 7.28 Hz, 2H, H2), 2.38 (s, 3H, H3), 3.56 (t, J = 6.96 Hz, 2H, H4), 7.21 (d, J = 7.29 Hz, 2H, H5), 7.62 (d, J = 8.12 Hz, 2H, H6), 8.23 (s, 1H, H7); 13C-NMR (CDCl3) δ (ppm): 11.85 (C1), 21.48 (C2), 24.09 (C3), 63.53 (C4), 127.98 (C5), 129.29 (C6), 133.70 (C7), 140.68 (C8), 160.77 (C9).
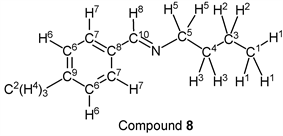
p-Tolualdehyde (1.20 g, 10.0 mmol) reacted with n-butylamine (0.80 g, 11.0 mmol) to give the corresponding pure compound (Compound 8) in 99% (1.74 g) as a clear oil in the same manner of procedure 2. All physical properties of this product were completely consistent with literature values [36] [41] or physical data of the commercially available compound: IR (neat): 500, 814, 1458, 1650, 2834, 2872, 2929, 2957 cm–1; 1H-NMR (CDCl3) δ (ppm): 0.92 (t, J = 7.36 Hz, 3H, H1), 1.36 (sext, J = 7.32 Hz, 2H, H2), 1.65 (quin, J = 7.28 Hz, 2H, H3), 2.35 (s, 3H, H4), 3.57 (t, J = 7.08 Hz, 2H, H5), 7.18 (d, J = 7.88 Hz, 2H, H6), 7.59 (d, J = 8.08 Hz, 2H, H7), 8.20 (s, 1H, H8); 13C-NMR (CDCl3) δ (ppm): 13.68 (C1), 20.21 (C2), 21.23 (C3), 32.80 (C4), 61.22 (C5), 127.73 (C6), 129.04 (C7), 133.48 (C8), 140.41 (C9) 160.43 (C10).
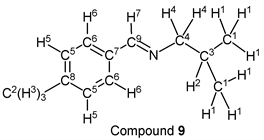
p-Tolualdehyde (1.20 g, 10.0 mmol) reacted with i-butylamine (0.80 g, 11.0 mmol) to give the corresponding pure compound (Compound 9) in 98% (1.71 g) as a clear oil in the same manner of procedure 2. All physical properties of this product were completely consistent with literature values [42] : IR (neat): 503, 811, 823, 1031, 1467, 1650, 2832, 2870, 2898, 2925, 2955 cm–1; 1H-NMR (CDCl3) δ (ppm): 0.94 (d, J = 6.68 Hz, 6H, H1), 1.99 (sep, J = 6.68 Hz, 1H, H2), 2.36 (s, 3H, H3), 3.40 (d, J = 6.64 Hz, 2H, H4), 7.19 (d, J = 7.88 Hz, 2H, H5), 7.60 (d, J = 8.09 Hz, 2H, H6), 8.18 (s, 1H, H7); 13C-NMR (CDCl3) δ (ppm): 20.68 (C1), 21.48 (C2), 29.57 (C3), 69.83 (C4), 128.01 (C5), 129.28 (C6), 133.74 (C7), 140.65 (C8), 160.77 (C9).
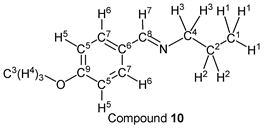
p-Anisaldehyde (1.37 g, 10.0 mmol) reacted with n-propylamine (0.65 g, 11.0 mmol) to give the corresponding pure compound (Compound 10) in 98% (1.73 g) as a yellow oil in the same manner of procedure 2. All physical properties of this product were completely consistent with literature values [43] [44] [45] or physical data of the commercially available compound: IR (neat): 503, 831, 1034, 1167, 1252, 1307, 1607, 1647, 2873, 2931, 2960, 2836 cm–1; 1H-NMR (CDCl3) δ (ppm): 0.93 (d, J = 7.40 Hz, 3H, H1), 1.70 (sext, J = 7.24 Hz, 2H, H2), 3.53 (t, J = 6.96 Hz, 2H, H3), 3.82 (s, 3H, H4), 6.91 (d, J = 8.76 Hz, 2H, H5), 7.66 (d, J = 8.80 Hz, 2H, H6), 8.19 (s, 1H, H7); 13C-NMR (CDCl3) δ (ppm): 11.88 (C1), 24.17 (C2), 55.35 (C3), 63.48 (C4), 113.95 (C5), 129.30 (C6), 129.55 (C7), 160.17 (C8), 161.45 (C9).
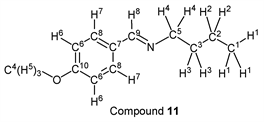
p-Anisaldehyde (1.37 g, 10.0 mmol) reacted with n-butylamine (0.80 g, 11.0 mmol) to give the corresponding pure compound (Compound 11) in 99% (1.89 g) as a pale yellow oil in the same manner of procedure 2. All physical properties of this product were completely consistent with literature values [46] [47] or physical data of the commercially available compound: IR (neat): 518, 832, 1034, 1167, 1252, 1607, 1649, 1512, 2835, 2872, 2931, 2957 cm–1; 1H-NMR (CDCl3) δ (ppm): 0.92 (t, J = 7.36 Hz, 3H, H1), 1.36 (sext, J = 7.32 Hz, 2H, H2), 1.65 (quin, J = 7.32 Hz, 2H, H3), 3.55 (t, J = 7.08 Hz, 2H, H4), 3.81 (s, 3H, H5), 6.90 (d, J = 8.76 Hz, 2H, H6), 7.64 (d, J = 8.76 Hz, 2H, H7), 8.17 (s, 1H, H8); 13C-NMR (CDCl3) δ (ppm): 13.92 (C1), 20.45 (C2), 33.10 (C3), 55.33 (C4), 61.39 (C5), 113.92 (C6), 129.30 (C7), 129.50 (C8), 160.04 (C9), 161.41 (C10).
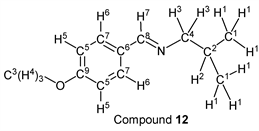
p-Anisaldehyde (1.37 g, 10.0 mmol) reacted with i-butylamine (0.80 g, 11.0 mmol) to give the corresponding pure compound (Compound 12) in 99% (1.89 g) as a yellow oil in the same manner of procedure 2. All physical properties of this product were completely consistent with literature values [48] [49] : IR (neat): 507, 832, 1033, 1167, 1253, 1512, 1607, 1649, 2836, 2870, 2900, 2928, 2955 cm–1; 1H-NMR (CDCl3) δ (ppm): 0.93 (d, J = 6.68 Hz, 6H, H1), 1.98 (sep, J = 6.68 Hz, 1H, H2), 3.38 (d, J = 6.68 Hz, 2H, H3), 3.82 (s, 3H, H4), 6.90 (d, J = 8.76 Hz, 2H, H5), 7.66 (d, J = 8.80 Hz, 2H, H6), 8.14 (s, 1H, H7); 13C-NMR (CDCl3) δ (ppm): 20.70 (C1), 29.62 (C2), 55.36 (C3), 69.79 (C4), 113.94 (C5), 129.34 (C6), 129.58 (C7), 160.19 (C8), 161.44 (C9).
3. Results and Discussion
i-Butylamine was used as a starting compound that had less reactivity, and we examined the effect of the equivalent amount of amine for solvent- and catalyst-free synthesis of the imine derivative (aldehyde: p-bromobenzaldehyde). Some results of this reaction were shown in Table 1. p-Bromobenzaldehyde reacted with 1.0 equivalent amount of i-butylamine for 3.0 h, the resulting reaction mixture was exposed to reduced pressure condition (>0.1 mmHg, 3.0 h) to give the corresponding imine (compound 6) in 92% yield (entry 1). In this reaction case, p-bromobenzaldehyde was recovered in 6%. This phenomenon meant that the synthetic reaction was not finished in these reaction conditions. Then, it was decided to investigate the reaction of p-bromobenzaldehyde using an excess of i-butylamine. When 1.1 equivalent amount of the amine was used as a nucleophile, the pure target compound was obtained in 99% yield without any purification (entry 2). Moreover, the desired imine compound was given, when a large excess of i-butylamine was used as a starting material. The pure target compound was obtained in excellent yield using 1.2, 1.3, 1.4 and 1.5 equivalent amounts of the amine (entries 3, 4, 5, and 6). In our previous work, we could
![]()
Table 1. Reaction of p-bromobenzaldehyde with i-butylamine.
find that 1.0 equivalent amount of i-butylamine reacted with benzaldehyde to give the corresponding imine in ca. 95% [24]. This fact suggested that the synthetic reaction using 1.0 equivalent of i-butylamine with benzaldehyde was not finished. Because we could find the same phenomenon in this investigation, it was clear that using slightly excess (1.1 eq.) of the amine having less reactivity was suitable for this solvent- and catalyst-free synthesis of the imine derivative. In reaction cases of entries 2, 3, 4, 5, and 6, we could not detect unreacted amine at all, because it was removed from the reaction mixture by the vacuum operation.
It was reported that the maximum yield of this imine synthesized from i-butylamine and p-bromobenzaldehyde (solvent: CH2Cl2, dehydrating agent: Na2SO4) was 88% in the past papers [39]. From this point of view, our synthetic method was considered to be superior.
We investigated some transformations of halogenated aromatic aldehydes to imine derivatives using alkyl amines (Table 2). When p-chlorobenzaldehyde was treated with n-propylamine (1.1 eq.) under solvent- and catalyst-free reaction conditions for 3.0 h followed by reducing pressure, the target imine (compound 1) was given in 98% yield (entry 1). The imine formation using n-butylamine as a nucleophile was carried out under the same reaction conditions, and the desired product (compound 2) was given, quantitatively (entry 2). Further, the sterically hindered amine derivative could be used as a substrate in this synthesis. When p-chlorobenzaldehyde reacted with i-butylamine without a solvent and a catalyst, the corresponding imine (compound 3) was obtained in excellent yield (entry 3). The tendency that the yield of the product was extremely high with any alkyl amine was also observed in the reaction of p-bromobenzaldehyde. For example, when n-propylamine and n-butylamine were employed as starting materials, target compounds (compounds 4 and 5) were obtained, quantitatively (entries 4 and 5). Moreover, p-bromobenzaldehyde reacted with i-butylamine smoothly, and the target imine was given in 99% yield (Table 1, entry 2).
![]()
Table 2. Reactions of halogenated aromatic aldehydes with alkyl amines.
There were reported that compounds 1, 2, 3, 4, and 5 could be synthesized by various reaction methods, previously. Compounds 1 and 2 were obtained in good yields from the aldehyde and the alkyl amine using microwave irradiation [34] [35]. Compounds 3, 4, and 5 were also synthesized from the corresponding aldehyde with alkyl amines in up to 96% yield, but heating, purification of the product, and reaction solvents (toluene or methanol) were needed to be carried out syntheses [36] [37] [38]. Compared to these cases, it was clear that our highly efficient synthetic method was suitable for syntheses of imine derivatives having halogen atom on their aromatic rings from the point of green chemistry. Because supplying energy from outside the reaction system, purification, and reaction solvents were unnecessary in our system.
Interestingly, it was found that the reaction was not completed in 3.0 hours when an aldehyde having less reactivity was used as an electrophile. p-Tolualdehyde and i-butylamine were chosen as model compounds, we investigated the reaction time of this synthetic method by thin-layer chromatography (TLC). When the reaction mixture was analyzed by TLC (silica gel, n-hexane/Et2O = 10/1) after 3.0 h, we found that this synthetic reaction was not finished. After 4.0 h, we could find the same phenomenon by TLC analysis. Therefore, we decided to carry out this type of reaction for 5.0 hours (Table 3). p-Tolualdehyde was treated with n-propylamine for 5.0 h followed by reducing pressure to give the corresponding imine derivative (compound 7) in 98% yield (entry 1). n-Butylamine and i-butylamine could be also used as nucleophiles in this reaction. p-Tolualdehyde was reacted with n-butylamine or i-butylamine to give the corresponding compounds (compounds 8 and 9) in 99% or 98% yields, respectively (entries 2 and 3). Furthermore, p-anisaldehyde having a strong electron-donating group (-OCH3) could be employed as a substrate. This aldehyde was transformed into the desired imine by use of n-propylamine, n-butylamine, or i-butylamine. Pure target imines (compounds 10, 11, and 12) were obtained in 98%, 99%, and 99% yields, respectively (entries 4, 5, and 6).
![]()
![]()
Table 3. Reactions of less reactive aromatic aldehydes with alkyl amines.
Compounds 7, 8, 9, 10, 11, and 12 had already been synthesized with another type of methods. Compounds 7, 8, and 10 were synthesized using a reaction solvent, a catalyst, and microwave irradiation, quantitatively [40] [41] [43]. In contrast, our method gave target compounds in excellent yields without these reaction factors. Whereas compound 9 was obtained in 96% yield when MgSO4 was used as a dehydrating agent [42], the same compound was given in 98% yield by our method. Compounds 11 and 12 were obtained in 90% and 95% yield [47] [48], but our method gave the same compounds quantitatively without a solvent and a catalyst. These facts indicated that our synthetic method of the alkylated imine derivative was better than past methods.
In 2019, we reported that active amines, such as aromatic amines and benzylamine were reacted with some aldehydes to give the desired imines in excellent yields [25]. In this previous reaction, the ratio of aldehyde/amine was 1/1 and reaction time was shorter than the reaction system that was reported in this paper. Against results of previous investigations, using 1.1 equivalent amounts of amines and the long reaction time (3.0 h or 5.0 h) were needed for finishing this synthesis. Then, we decided to elucidate the reason why such a phenomenon was found by computational chemistry. First, we compared formation energies of the benzylated imine derivative with that of n-propylated, n-butylated, and i-butylated imine derivatives. We calculated formation energy differences between imines and p-bromobenzaldehyde, and used them as indexes of stability of imine derivatives. Results were shown in Table 4. Calculation data indicated that all imine derivatives were more stable than p-bromobenzaldehyde. Formation energies of the benzylated imine (entry 1) were lower than that of alkylated imines (entries 2, 3, and 4) in all the calculation methods (HF/6-31+G*, HF/6-311G*, ωB97X-D/6-31G*, EDF2/6-31G*). These calculation results showed that the formation of the benzylated imine was easy to progress, because of the low formation energy of the target compound.
Next, we tried to calculate energy levels of molecular orbitals of amines and aldehydes. Energy levels of HOMO (Highest Occupied Molecular Orbital) or LUMO (Lowest Unoccupied Molecular Orbital) of various compounds were summarized in Table 5.
![]()
![]()
Table 4. Comparison of the formation energy of p-bromobenzaldehyde and the corresponding imine.
a)ΔE = (Formation energy of imine)− (Formation energy of aldehyde).
![]()
![]()
Table 5. Energy levelsa) of molecular orbitals of amines and aldehydes.
a)Calculation method = ωB97X-D/6-31G*.
In general, it was well known that molecules with higher HOMO energy levels had higher nucleophilicity, and molecules with lower LUMO energy levels had higher electrophilicity. The HOMO energy level of benzylamine was higher than that of n-propylamine, n-butylamine, i-butylamine (entry 1 vs. entries 2, 3, and 4). These calculation results suggested that the synthetic reaction using benzylamine progressed fast because of its strong nucleophilicity. It had become clear that reactions of alkyl amines needed strong reaction conditions (aldehyde/amine = 1/1.1, and the long reaction time) because of high formation energies of target compounds and low nucleophilicity of starting amines. This calculation of the energy level also revealed the reason for the long reaction time of cases of less reactive aldehydes. LUMO energy levels of p-tolualdehyde and p-anisaldehyde were higher than that of halogenated aromatic aldehydes (entries 5 and 6 vs. entries 7 and 8). It was meant that p-tolualdehyde and p-anisaldehyde had low electrophilicity. Due to this low electrophilicity, we thought that reactions of p-tolualdehyde or p-anisaldehyde required the long reaction time. As described above, the property of this reaction could be clarified using computational chemistry. In the future, we will study further reaction mechanisms by the identity of the reaction intermediate and using computational chemistry.
4. Conclusion
In summary, we developed the effective synthetic method of the imine derivative using a weak nucleophile (alkyl amine) under solvent- and catalyst-free conditions. In this reaction system, it was found that pure target compounds could be obtained in excellent yields by using 1.1 equivalents of alkyl amines to aldehydes and the long reaction time compared with our previous work. When aldehydes having electron-withdrawing groups were used as starting compounds, reactions were completed in the short reaction time (3 h + 3 h), whereas the reaction time was prolonged (5 h + 3 h) by use of aldehydes having electron-donating groups. No purifications of target compounds were required because all unreacted compounds were removed by the reduced pressure operation. The reaction property of this synthetic system was elucidated by computational chemistry. When a weak nucleophile was used, the formation energy of the target product was high, and it was indicated that the progression of this reaction was somewhat disadvantageous. Energy levels of HOMO of amines were compared with each other, and then it was revealed that alkyl amines were less reactive nucleophiles with their low energy levels. Furthermore, it was found that aldehydes having electron-withdrawing groups were superior electrophiles because of their lower energy levels of LUMO. In the future, we will study the intermediates of this reaction in detail to clarify the reaction mechanism.