Photophysical Properties of Perylenetetracarboxylic Diimide Dimers with Slipped “Face-to-Face” Stacked Structure and Different Bay Substitutions ()

1. Introduction
In natural light-harvesting systems, a slipped arrangement of chlorophyll dyes is accomplished by proteins or metal-ion coordination, providing J-type aggregates [1]. To mimic natural archetype, many chromophores were constructed into J-aggregates by self-assemble process. Among these dyes, porphyrin dyes [2] are the most popularly investigated organic dyes because of their similar molecular structure and photophysical properties with natural bacteriochlorophyll a (Bchl a) molecules [3] [4]. However, since these chromophores exhibit much less favorable optical properties, especially a rather weak photoluminescence resulting from an almost forbidden lowest-energy transition, porphyrin aggregates can compete in this regard neither with their natural chlorophyll counterparts, nor with cyanine dye-based synthetic J-aggregates. Other than porphyrin dyes, perylenetetracar- boxylic diimides (PDIs) [5] are desirable building blocks for novel functional J-type aggregates due to their outstanding photophysical properties, in particular the exceptional high fluorescence quantum yields and excellent stabilities towards heat and light.
Several J-type aggregates assembled from PDI based on non-covalent intermolecular interactions, such as hydrogen bonding and/or π − π stacking interaction, have been intensively studied [6]-[8]. However, these aggregates showed significant differences on photophysical properties. And it is very difficult to establish direct correlation between the molecular structure and optical properties by self-assembled PDI aggregates due to the flexible structure of the supramolecular system. So the covalent linked J-aggregate with rigid structure is necessary. And Wasielewski group has reported three covalently linked core-unsubstituted PDI dimers with slip- stacked structure [9]. In addition, a series of covalently linked core-substituted PDI dimers and trimers with diphenoxyl groups have also been reported by our group [10]. In both of these studies, the degree of slipping of the two PDI chromophores is tuned by changing the spacer, which can affect the photophysical property of J-aggregate. However, we can also control the degrees of slipping and/or rotation of the two PDI rings relative to each other by introducing different substitute groups to the bay area of PDI ring as revealed by the previous literatures [11].
Herein, we report the design, synthesis and optical properties of six PDI dimers with different substituents at the bay positions of the PDI ring (Scheme 1, 4-9). The difference on the number or species of side groups attached at the bay positions of the PDI rings in these dimers induces different degrees of slipping and/or rotation of the two PDI rings relative to each other, which can lead to different electronic coupling between the two PDI subunits in these compounds.
2. Experimental Section
2.1. General Information
1H NMR spectra were recorded on a Bruker 300 or 400 MHz NMR spectrometer with chemical shifts reported in ppm (TMS as internal standard). MALDI-TOF mass spectra were recorded with a Bruker/ultra flex instrument. Absorption spectra and fluorescence spectra were measured on SHIMADZU UV-2450 spectrophotometer and Edinburgh Instruments FLS920 spectrophotometer, respectively. The fluorescence lifetimes were measured on FLS920 with time correlated single photon counting (TCSPC) method by excitation with a 400 nm picosecond laser (EPL 405) operated at 10 MHz. Fluorescence quantum yields were calculated with N,N’-di(n-hexyl) perylene-3,4:9,10-tetracarboxylic diimide (100%) as standard.
2.2. Materials
Compounds 1-5, 10-13 were prepared following the published procedures [10] [12]-[16]. All other chemicals are purchased from commercial source and used as received without further purification.
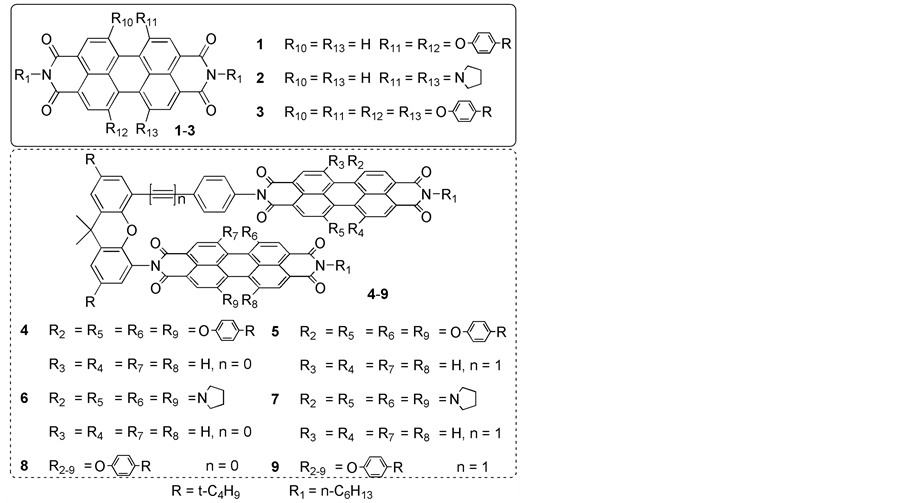
Scheme 1. Structures of the compounds 4-9 and the reference compounds 1-3.
Compound 6: A mixture of 12 (42.8 mg, 0.1 mmol), 11 (122.6 mg, 0.2 mmol), and imidazole (0.5 g, 7.4 mmol) in toluene (50 mL) was refluxed under N2 for 12 h. After the solvent was evaporated under reduced pressure, the residue was dissolved in chloroform and washed with water for several times to remove imidazole. The chloroform was then removed under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate/chloroform = 1/19). After recrystallization from chloroform and methanol, compound 6 was collected as a green solid (17.8 mg, 11%). M.p. > 300˚C; 1H NMR (300 MHz, CDCl3, 25˚C, TMS): 8.67 (d, 1H), 8.61 (s, 1H), 8.57 (s, 1H), 8.47 (d, 1H), 8.12 (d, 1H), 8.04 (d, 1H), 8.01 (s, 1H), 7.90 (s, 1H), 7.72 (d, 1H), 7.59 (s, 1H), 7.55 - 7.52 (m, 2H), 7.44 (s, 1H), 7.38 (d, 2H), 7.28 (d, 1H), 7.20 (s, 1H), 7.13 (s, 1H), 6.10 (d, 2H), 4.29 (t, 2H), 3.91 - 3.51 (m, 12H), 2.82 - 2.61 (m, 6H), 1.97 (m, 16H), 1.85 (m, 5H), 1.75 (s, 3H), 1.52 (m, 2H), 1.40 (m, 13H), 1.33 (m, 9H), 1.30 - 1.0 (m, 8H), 0.94 (t, 3H), 0.84 (t, 3H); MS (MALDI-TOF): m/z: calcd: 1618.78; found: 1619.54 [M + H+]; Elemental analysis (%), calculated for C105H102N8O9: C 77.85, H 6.35, N 6.92; found: C 77.62, H 6.41, N 7.05.
Compound 7: By using a similar procedure to that for preparing compound 6, with 13 (45.2 mg, 0.1 mmol) instead of 12 as starting material, compound 7 was synthesized. The product was purified by column chromatography on silica gel (ethyl acetate/chloroform = 1/19). After recrystallization from chloroform and methanol, compound 7 was collected as a green solid (21.4 mg, 13%). M.p. > 300˚C; 1H NMR (400 MHz, CDCl3, 25˚C, TMS): 8.61 (s, 1H), 8.57 (s, 1H), 8.56 (d, 1H), 8.46 (d, 1H), 8.30 (s, 1H), 8.27 (s, 1H), 8.19 (d, 1H), 8.14 (d, 1H), 7.91 (d, 1H), 7.76 (d, 3H), 7.58 (s, 1H), 7.30 (s, 1H), 7.23 (s, 1H), 6.92 (s, 1H), 6.34 (d, 2H), 6.16 (d, 2H), 4.25 (t, 2H), 3.82 - 3.40 (m, 12H), 2.98 - 2.89 (m, 6H), 2.70 - 2.54 (m, 4H), 2.10 (m, 16H), 1.77 - 1.73 (m, 8H), 1.52 (m, 2H), 1.40 (m, 13H), 1.29 (m, 9H), 1.09 - 1.05 (m, 4H), 0.92 (t, 3H), 0.73 (t, 3H); MS (MALDI-TOF): m/z: calcd: 1642.78; found: 1643.81 [M + H+]; Elemental analysis (%), calculated for C107H102N8O9: C 78.17, H 6.25, N 6.82; found: C 78.52, H 6.11, N 6.55.
Compound 8: By using a similar procedure to that for preparing compound 6, with 10 (213.6 mg, 0.2 mmol) instead of 11 as starting material, compound 8 was synthesized. The product was purified by column chromatography on silica gel (petroleum ether/chloroform = 1/2). After recrystallization from chloroform and methanol, compound 8 was collected as a red solid (22.8 mg, 9%). M.p. > 300˚C; 1H NMR (300 MHz, CDCl3, 25˚C, TMS): 8.12 (s, 2H), 8.01 (s, 2H), 7.93 (s, 2H), 7.61 (s, 2H), 7.50 (s, 2H), 7.36 (s, 2H), 7.29 (d, 6H), 7.17 - 6.91 (m, 18H), 6.75 (d, 4H), 6.63 (d, 4H), 6.23 (d, 2H), 4.10 (t, 2H), 3.20 (t, 1H), 2.54 (t, 1H), 1.81 (s, 3H), 1.65 (t, 2H), 1.60 (s, 3H), 1.36 - 1.16 (m, 104H), 0.86 (t, 3H), 0.79 (t, 3H); MS (MALDI-TOF): m/z: calcd: 2527.26; found: 2528.97 [M + H+]; Elemental analysis (%), calculated for C169H170N4O17: C 80.26, H 6.77, N 2.22; found: C 80.13, H 6.65, N 2.52.
Compound 9: By using a similar procedure to that for preparing compound 7, with 10 (213.6 mg, 0.2 mmol) instead of 11 as starting material, compound 9 was synthesized. The product was purified by column chromatography on silica gel (petroleum ether/chloroform = 1/2). After recrystallization from chloroform and methanol, compound 9 was collected as a red solid (25.5 mg, 10%). M.p. > 300˚C; 1H NMR (300 MHz, CDCl3, 25˚C, TMS):
8.13 (s, 4H), 8.11 (s, 1H), 8.07 (s, 1H), 7.56 (s, 2H), 7.47 (s, 1H), 7.22 - 7.13 (m, 11H), 7.08 - 6.91 (m, 12H), 6.78 (d, 4H), 6.72 - 6.58 (m, 10H), 6.42 (d, 2H), 4.09 (t, 2H), 3.27 (m, 1H), 2.69 (m, 4H), 2.46 (m, 1H), 1.71 - 1.64 (m, 8H), 1.34 - 1.19 (m, 100H), 0.86 (t, 3H), 0.76 (t, 3H); MS (MALDI-TOF): m/z: calcd: 2551.26; found: 2552.24 [M + H+]; Elemental analysis (%), calculated for C171H170N4O17: C 80.44, H 6.71, N 2.19; found: C 80.68, H 6.75, N 2.31.
3. Results and Discussion
3.1. Molecular Design and Synthesis
It has been proved that introducing different substituents at the bay positions of the PDI ring is an efficient way to change photophysical properties of PDI compounds [17]. Therefore, different numbers of phenoxy or pyrrolidinyl groups are incorporated into the bay positions of PDI rings. And the different side groups attached at the bay positions of the PDI rings induce different degrees of slipping and/or rotation of the two PDI rings relative to each other, which can change the interactions between the two PDI rings. The synthetic procedures are shown in Scheme 2. All the new compounds were fully characterized with 1H NMR, MALDI-TOF mass spectra, as well as elemental analysis.
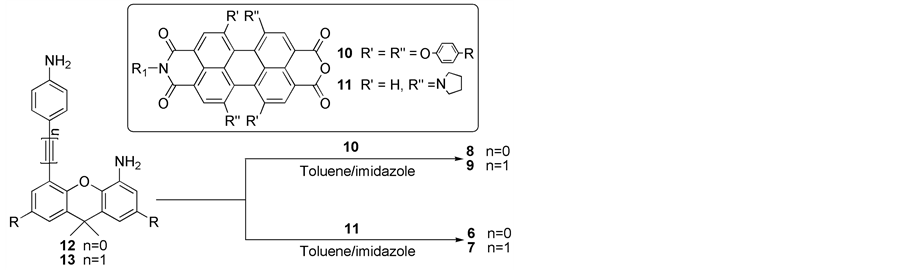
Scheme 2. Synthesis of the compounds.
3.2. Uv-vis Absorption Spectra
Figure 1 shows the electronic absorption spectra of dimer 4-9 and those of their monomer 1-3 in toluene.
Dimer 4 presents two absorption bands at 550 nm and 518 nm (Figure 1(a)). The former red-shifted by 11 nm while the latter blue-shifted by 21 nm compared to the maximal absorption band of monomer 1. Notably, the intensities of these two bands are nearly the same. Two absorption bands at about 549 nm and 515 nm are observed in the absorption spectrum of dimer 5. The maximal absorption band is red-shifted by about 10 nm compared with that of monomer 1 [10].
As shown in Figure 1(b), the maximum absorption band of dimer 6 appears at 688 nm, which red-shifted only 2 nm in comparison with that of monomer 2. Slight enhancement of the 0 - 1 vibronic band can be identified from the normalized absorption spectra (the inset of Figure 1B). These results suggest that the interaction between the two PDI rings in dimer 6 is significantly weaker than that in dimer 4. The absorption spectrum of dimer 7 is similar in shape with that of monomer 2. But the maximal absorption peak of dimer 7 red-shifted by 12 nm with respect to that of monomer 2. This is a typical feature of J-type aggregates, which is also observed in linear PDI aggregates [19].
The absorption spectrum of dimer 8 is characterized by the maximal absorption peak at about 584 nm (Figure 1(c)), which red-shifted 12 nm with respect to that of monomer 3, corresponding well with the “J” type structure of dimer 8. This red-shift of the absorption maximum of dimer 8 is much larger than that of dimer 6, which implies that the interactions between the two PDI units in dimer 8 is stronger than that in dimer 6. Dimer 9 presents the maximal absorption band at 583 nm, which red-shifted by 11 nm with respect to that of monomer 3. It is surprising that the absorption spectrum of dimer 9 is similar with that of dimer 8 based on the relative large difference between the absorption spectra of dimer 4 and 5 as well as 6 and 7. This can be attributed to the effect of the four bulky phenoxyl groups at the bay positions of the PDI ring. The larger displacement between the two PDI rings in dimer 9 with respect to that in dimer 8 has decreased the steric hindrance in somehow and then the neighboring two PDI rings can take a relative more parallel orientation. This more parallel conformation has enforced the interactions between the two PDI rings. However, the larger displacement along the long axis of the PDI ring leads to larger center-to-center distance and smaller interactions between the two neighboring PDI rings. The result of these two controversial effects is the similar absorption spectra of dimer 9 with that of dimer 8.
The maximal absorption bands for dimer 5, 7, and 9 red-shifted by 10 nm without obvious change on shape relative to their corresponding monomer 1, 2, and 3. As discussed in dimer 4, 6 and 8, different red-shifts or absorption spectra shapes should be observed because of the different steric hindrance from various substituent groups at bay positions in dimer 5, 7 and 9. However, due to the large longitudinal displacement of dimer 5, 7 and 9, the center-to-center distance between the PDI rings (L) increased significantly. According to the exciton theory, the exciton splitting is proportional to the value of L−3 [18]. The larger center-to-center distance makes the interactions between the two PDI subunits quite weak. Therefore, the effect on the absorption spectra of dimer 5, 7 and 9 resulted from different substituent groups at bay positions can be negligible.
3.3. Fluorescence Spectra and Fluorescence Lifetime
Figure 2 shows the fluorescence spectra of dimer 4-9 and their monomer 1-3 in toluene. Notably, weak emis-
![]()
Figure 1. Absorption spectra of dimer 4-9 and of their monomer 1-3 in toluene (10−5 M). The inset shows the normalized (at maximum) absorption spectra.
![]()
Figure 2. Fluorescence spectra of dimer 4-9 and of their monomer 1-3 in toluene (10−6 M). The inset shows the normalized fluorescence spectra.
sions were detected for both dimer 6 and 7, which is different from the previously reported “H” type dimer [20b]. Due to the charge separation between the two PDI units in a “face-to-face” stacked PDI dimer, the fluorescence is quenched completely. The fluorescence properties of these compounds are summarized in Table 1.
As shown in Figure 2(a), the emission maximum of dimer 4 appears at 628 nm, which red-shifted by 59 nm with respect to that of monomer 1. An emission band at about 584 nm is observed in the emission spectrum of dimer 5, which red-shifted by 15 nm compared with that of monomer 1. According to the previous report, the emission of dimer 4 and 5 can be attributed to the “excimer-like” states [10]. It is worth noting that the emission from “excimer-like” states of PDI varied dramatically for PDI dimers with different stacked structures. The difference between the emission of the “excimer-like” states of dimer 4 and 5 indicates that large displacement along the molecular long axis will cause a short wavelength emission for the “excimer-like” states.
The emission spectra of dimer 6 and 7 show the emission band at 728 nm and 725 nm (Figure 2(b)), which red-shifted by only 8 and 5 nm with respect to that of monomer 2, respectively. The emission of dimer 6 and 7 seemingly origins from their monomer if only in terms of the small red-shift and similar emission spectra shape. However, the fluorescence quantum yields of dimer 6 and 7 are measured to be 0.1% and 0.8%, respectively, which drop a lot compared to that of monomer. Moreover, the fluorescence of both dimer 6 and 7 decay two-exponentially and two fluorescence lifetimes are measured for both of them from the fitting of the fluorescence decay curves, which is not observed in monomer 2. All results indicate that the emission bands at 728 nm and 725 nm of dimer 6 and 7 are not monomeric emission.
The emission maximum of dimer 8 appears at 620 nm with a quantum yield of 21.2% and lifetime of 7.8 ns. The emission wavelength is red-shifted by 17 nm relative to that of monomer 3, and the fluorescence quantum yield drops to almost one third of the monomer, which is obviously different from that observed for the typical “J-type” aggregates [6] [7]. The fluorescence lifetime increases slightly from 6.1 ns for the monomer 3 to 7.8 ns for dimer 8. The Stokes shift of the emission band is 36 nm, which is also significantly larger than that observed for the typical “J-type” aggregates [6]-[8]. All the characteristics of the emission of dimer 8 as mentioned above suggest that the 620 nm emission can be assigned to the “excimer-like” state of dimer 8. However, because of the weak interactions between the two PDI rings in dimer 8, the characteristics of the emission of dimer 8 are slightly different from that observed for the classic “H-type” aggregates [20]. In addition, the 17 nm red-shift on the maximal emission band of dimer 8 with respect to that of the monomer 3 is smaller than that of dimer 4 (59 nm), but larger than that of dimer 6 (8 nm). This indicates that the interactions between the two PDI rings in dimer 8 is weaker than that in dimer 4, but stronger than that in dimer 6, which agrees well with the result of the absorption spectra. The emission spectrum of dimer 9 presents an emission band at about 616 nm which is red-shifted 13 nm relative to that of monomer 3. This emission has a quantum yield of 39.5%. The fluorescence lifetime of dimer 9 is 7.21 ns, which can be similarly attributed to the emission of “excimer-like” states.
Unlike the similar absorption spectra of dimer 5, 7 and 9, significantly different red-shifts on the maximal
![]()
Table 1. The fluorescence properties of the compounds in toluene.
[d]From ref 10.
emission band relative to their monomeric counterparts have been observed in the fluorescence spectra of them. This suggests that the fluorescence spectra are much more sensitive to the structure variation than the absorption spectra. In another word, the effect of the substituents on the fluorescence spectra of dimer 5, 7 and 9 is significant. In addition, the fluorescence quantum yields of dimer 5, 7 and 9 are higher than those of their corresponding dimer 4, 6 and 8. This is because the weaker interactions between the two PDI units in these compounds caused by the larger displacement along the long axis of the PDI ring.
4. Conclusion
In summary, a series of covalently linked perylenetetracarboxylic diimides (PDIs) dimers with slipped “face-to- face” stacked structure and different substituents at the bay positions were designed and prepared. Even though these dimers are J-aggregates in structure, the fluorescence spectra of them are all dominated by the emission of “excimer-like” states. The properties of this “excimer-like” emission are determined by the structure and the interactions between the PDI subunits of these dimers. Weak interactions between the PDI subunits will lead to an “excimer-like” emission which similar with that of monomeric fluorescence.
Acknowledgements
XL thank the Natural Science Foundation of China (Grand No. 21073112, 21173136, and 91233108), the National Basic Research Program of China (973 Program: 2012CB93280). HL thanks the Fundamental Research Funds for the Central Universities (15CX05067A) and Qingdao postdoctoral fellowship program (2015245).