1. Introduction
Tetrahydrofuran (THF) is a small cyclic ether frequently adopted as solvent, thanks to its remarkable physicochemical properties (see Figure 3 for the molecular structure). The molecule of THF is a very interesting model for studies related to structure of organic rings, torsional barriers and vibrational modes. A series of reported results point out that the molecule presents pseudo-rotation, which consists in the transition from the Cs (envelope) to the C2 (twist) conformation [1] -[3] with the last one being the most stable of them [4] .
As a structural unity of many carbohydrates, it is a precursor of biologically active molecules [5] . Lots of important natural substances contain the THF structure in their molecules as, for example, marine macrolides and tetrahydrofuran lignans, just to name two that are subject of study in our laboratory. Marine macrolides are secondary metabolites with important biological activities as a defense mechanism to the aggression of the environment. Because of that they are excellent candidates for the investigation of new bioactive molecules with high pharmacological potential [6] [7] . Some of them have reached the clinical trial stage or the market, as is the case of the anticancer agent eribulin, the analogue of the macrolide halicondrin B [8] [9] . On the other hand, some tetrahydrofuran lignans have showed a broad range of biological activities, including anti-inflammatory, analgesic, trypanocidal, schistosomicidal, antileishmanial and antimalarial [10] - [15] . No less important is that THF molecule can be used as an analogous of the DNA skeleton, since DNA can be viewed as a chain consisting of various THF molecules linked each other by phosphate ions. This model has been used, for example, in studies on DNA mutations induced by ionizing radiation [16] .
Liquid THF does not spontaneously produce solvated electrons on solvation of alkali metal atoms such as sodium [17] . It is thought to allow the creation of larger volumes in which free electrons can be accommodated [18] [19] . Because of that, it has recently been used as a novel solvent in investigations of solvated electron that are of importance in improving our understanding of electron-transfer reactions and radiation chemistry [17] [18] .
Most of the current understanding of THF liquid structure is almost exclusively derived from computer simulation studies [20] - [24] . All these investigations have been parameterized to reproduce reasonably macroscopic physical properties such as the liquid’s diffusion coefficient or specific heat capacity. Because of that the resulting structure is entirely dependent upon the resulting interatomic and intermolecular force fields and conclusions concerning molecular interactions and local structure are largely speculative [25] . Recently, we began a systematic study on the structure of liquids, both pure [26] as mixtures [27] [28] , using neutron diffraction with isotope substitution and EPSR simulation [29] - [31] . In a previous application of that methodology Bowron et al. have investigated the structure of liquid THF detailing the molecular correlations and the void structure in the liquid [25] . The present study comes on the heels of those previously cited aiming to deep the knowledge on the structure of liquid THF. For that, the classic Monte Carlo approach used in the EPSR methodology was applied to equilibrate the liquid and the simulation box explored using a protocol developed in our group [32] . The results complement those reported previously and help to have a more detailed view of the structure of the liquid.
2. Methodology
2.1. Molecular Models and Intermolecular Potential Function
The molecular geometry used for the THF molecule has been that derived previously from neutron diffraction with EPSR methodology [25] . In the present simulations have been used the intermolecular potential optimized for Chandrasekhar and Jorgensen [20] [21] . The molecules are all-atoms represented and were maintained rigid during the simulation which is a quite appropriated approximation for THF, as it has been reported [20] [21] . Of course, this approach introduces uncertainties in the simulations which can be significant if one studies strongly conformer-dependent properties such as molecular polarization for example, which it is not the case here.
As it is classically performed in this kind of simulations, the energy Eab between the interacting sites in the molecules a and b is calculated by a sum of Coulomb and Lennard Jones potentials centered on the sites:
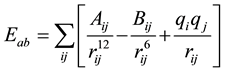 (1)
(1)
here, rij is the distance between site i in a and site j in b and qi and qj are fractional charges located on the i and j molecular sites. For each site k, the parameters Akk and Bkk were given by  and
and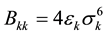 , where
, where 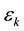 and
and 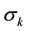 are the Lennard-Jones parameters for the kth site. Parameters Aij and Bij for non-diagonal interactions
are the Lennard-Jones parameters for the kth site. Parameters Aij and Bij for non-diagonal interactions 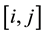 were obtained using the geometric combining rules
were obtained using the geometric combining rules 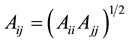 and
and 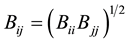 [33] . As it is easily realized, this is a pairwise potential where Eab is the energy between the molecular pairs.
[33] . As it is easily realized, this is a pairwise potential where Eab is the energy between the molecular pairs.
2.2. Monte Carlo Simulations
The simulations were carried out in the NPT ensemble at 298 K and 1 atm, with Metropolis importance sampling and periodic boundary conditions [33] on systems consisting of a cubic box containing 500 molecules. In the calculation of the total configurational energy using Equation (1), a full intermolecular interaction was considered whenever the rij site-to-site distance gets inside a cutoff radius of 11 Å. Considering that for neutral molecules the main contribution to the cohesive energy comes from the dipole-dipole interaction, which decrease asymptotically with 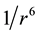 [34] , long-range interactions beyond the cutoff radius were not considered. The use of approaches like Ewald summation, for example, also very used in our laboratory, does not introduce any gain in the results [35] [36] .
[34] , long-range interactions beyond the cutoff radius were not considered. The use of approaches like Ewald summation, for example, also very used in our laboratory, does not introduce any gain in the results [35] [36] .
3. Results and Discussion
It is not the aim of this work to analyze the radial distribution functions g(r) obtained, since they were detailing analyzed previously [25] . However, just to facilitate the discussion and give a first view on the liquid, it is presented in Figure 1 the pair distribution functions for the heavy atoms obtained from the simulations.
It is clear from the curves that THF is a liquid poorly organized. All radial distribution functions started about 3 Å with very broad curves (some correlations with methyl hydrogen atoms are somewhat shorter), reflex of the weak interaction among molecules. Accordingly, Borow et al. reported a coordination number of 12.6 for the distribution of the molecular centers, which is the typical coordination number for the distribution of hard spheres [25] . The interactions between the molecules are structured by the oxygen and the C(O) sites. Furthermore, the interactions at the non-polar part of the molecule are driven by the general molecular packing within the liquid, as it has been discussed [25] . Typical preferred orientation of molecules has been explored through intermolecular orientational correlation maps (OCM) [25] . The authors shown there are three preferred pairs formed in the liquid, all of them T-like with the central molecule pointing into the center of a neighboring THF molecular ring. However, no information is given on the stability of these pairs. Thus, to have an idea about that stability, the pair energy distribution along the simulation has been accumulated which result is shown in Figure 2 (left).
Differently of other liquids like acetone and N-methylformamide (NMF) [37] or dimethylsulfoxide (DMSO) and water [38] , which present an structured pair energy distribution, consequence of existence of pairs with different stability in the liquids, for THF the pair energy distribution curve show no structure. There is a unique band of energy, indicating that the energy relative to the different pairs corresponding to the OCM is encom- passed for that band. Accordingly, the three molecular pairs reported are very similar [25] . Furthermore, the range of energy distribution (−3 - 0 kcal∙mol−1) is less negative than that observed for NMF or DMSO, for example [37] [38] , indicating weak intermolecular interaction in THF, agreeing with the absence of structure suggested based in the g(r) plots. The mono-modal curve observed in Figure 2 is straight in line with the single peak structure of molecular centers distribution function for THF reported [25] . These results are reflected directly in the absence of any trace of double layer in the liquid, as it is clear from the graph shown in Figure 2 (right).
Hence, from the OCM were derived three preferential T-like molecular pairs [25] which have been showing here are encompassed by a single band of energy. However, although the T-like configurations are the most favorable, the OCM suggest that it is possible for THF molecular rings to interact via antiparallel face to face approach [25] . Having in mind this statement, the simulation box was explored looking for this type of correlation pairs. The search was carried out at well-defined ranges of distance, in order to detail the different types of pair in each region as much as possible. Figure 3 shows three parallel pairs obtained as a function of the interval of distance together with some common geometric parameters.
The results show that, effectively there are parallel pairs in the liquid however they have a tendency to change from completely antiparallel to less antiparallel at larger distances. Concomitantly, as the distance increase, they tend to the T-like form. These are interesting results, since they permit to have a better insight on the behavior of the antiparallel pairs that have been proposed previously [25] . On the other hand, even though the diversity of pairs found in the solvent, the geometric parameters shown are roughly the same for all of them, consequence of the change in the relative position of the molecules. Accordingly, these values rather agree with the distances of the peaks seen in the g(r) plots of Figure 1. Thus, the liquid is effectively constituted of T-like and antiparallel pairs, with the last ones changing from antiparallel to parallel as a function of the distance between the molecules and tending to the T-like for larger distances. Note that this result does not mean that the T-like pairs exist only at larger distances, but that the antiparallel pairs, that are most stable at short distances, tend to change for T-like as the intermolecular distance increase. The presence in the solvent of all these types of molecular pairs may implicate in, or it is a consequence of the poor packing of the THF.
![]()
![]()
Figure 1. Intermolecular site-site radial distribution functions derived from the simulations for THF at room temperature. O = oxygen, C(O) = carbon bonded to oxygen, C = carbon bonded to C(O).
![]()
![]()
Figure 2. Pair energy distributions derived by the simulation for THF molecules in the liquid (the curves were truncated at 7 for plotting) (left); Number of neighbors as a function of distance between the molecules (right).
![]()
![]()
![]() (a) (b) (c)
(a) (b) (c)
Figure 3. Parallel pairs as a function of the intermolecular distance in the liquid. (a) 3 - 4 Å; (b) 4 - 6 Å; (c) 6 - 7.5 Å. Some geometric parameters common to the pairs: r(O∙∙∙C(O)) = 3.53 Ǻ, r(O∙∙∙O) = 5.26 Ǻ, r(C(O)∙∙∙C(O)) = 4.66 Ǻ, r(C(O )∙∙∙C) = 6.22 Ǻ.
The relative positioning of molecules in the pairs may be even clearer, observing the relative positioning of the dipole moment vectors as a function of the distance, result that is shown in Figure 4. It is clear from the graphic that the molecules in the pairs are antiparallel to molecular distances between 3 and 4 Å, with the angle between the dipole moment vectors about 130˚ and that the angles tend to values about 90˚ for larger molecular distances. These results agree quite well with the whole molecular analyses discussed here and previously as well [25] .
![]()
Figure 4. Average dipole-dipole correlation as a function of oxygen- oxygen distance. θ is the angle between the dipole moments of the two THF molecules considered.
4. Conclusion
Liquid THF is a popular solvent, thanks to its remarkable physicochemical properties, besides being a structural unity of many important natural substances. It is also thought to allow the creation of larger volumes in which free electrons can be accommodated. Because of that, it has recently been used as a novel solvent in investigations of solvated electrons. The structure of the liquid has been explored via Monte Carlo simulations in the NTP ensemble having a previous EPSR study as starting point. The results point to the existence of different types of pairs, changing from antiparallel to T-shape, depending on the distance between the molecules. It is suggested that this diversity of pairs is closely related with the absence of packing and the formation of voids in the liquid.
Acknowledgements
A.B. thanks CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior) for the concession of a grant that help supporting this work.
NOTES
*Corresponding author.