Solvent Free One-Pot Synthesis of 1,2,4,5-Tetrasubstituted Imidazoles Catalyzed by Secondary Amine Based Ionic Liquid and Defective Keggin Heteropoly Acid ()
1. Introduction
Tetrasubstituted imidazoles are important heterocycles with wide biological applications. Their synthesis and reactions form a significant part of the study of heterocycles with medicinal use [1]. Tetrasubstituted imidazole scaffold is the most active constituent in many biological systems and drug molecules such as olmesartan medoxomil, losartan, eprosartan and trifenagrel [2-4] as well as other natural products of pharmaceutical importance [5-7]. Multicomponent reaction (MCR) is a powerful tool in generating molecules with diverse functionality in a single synthetic protocol [8-10] and forms the basis of the synthesis of 1,2,4,5-tetrasubstituted imidazoles. Important synthetic methods include one-pot thiazolium catalyzed addition of an aldehyde to an acyl imine followed by ring closure to the imidazole [11], condensation of arylglyoxal, 1˚-amine, carboxylic acids and isocyanates on Wang resin followed by cyclization in the presence of acetic acid [12], by a hetero-Cope rearrangement [13], a direct synthesis from alkenes involving two step ketoiodonation/cyclization protocol [14], Domino reaction of 2-azido acrylates and nitrones [15]. Other synthetic methods include the condensation of benzoin and benzoin acetate with aldehyde, 1˚-amine, ammonia in the presence of copper acetate [16], one-pot synthesis using BF3-SiO2 as catalyst [17], cyclization of sulphonamide with mesoionic 1,3-oxazolinium-5-olates [18], condensation of β-carbonyl-N-acyl-N-alkylamines with NH4OAc in refluxing acetic acid [19,20], conversion of N-2-oxoamides with ammonium triflate [21] besides others [22-25]. Several catalysts have been explored for performing the four components’ reaction to give the target product notably the use of L-proline [26], zeolite HY and silica gel [27], Keggin type heteropolyacid [28], LaCl3 catalyzed synthesis using urea as a source of ammonia [29]. Shaterian et al. reported the synthesis of tetrasubstituted imidazole used Brønsted acidic ionic liquid, N-methyl-2-pyrrolidonium hydrogen sulphate, as catalyst, but long reaction time, high amount of catalyst used and the time-consuming procedure for the preparation of the IL (12 hrs) are some of the disadvantages [30]. Most of the procedures reported are associated with one or more disadvantages such as use of expensive reagents and catalysts, mineral acids and requirement of large amount of catalysts which eventually results in generation of toxic wastes, longer reaction time and tedious work-up.
2. Results and Discussion
In a broad programme of developing efficient, selective and eco friendly synthetic methods for pharmacologically important heterocycles, we explored the applicability of green promoters namely the simple acidic ionic liquids (IL) as well as the defective Keggin type heteropolyacids (HPA) in the synthesis of 1,2,4,5 tetrasubstituted imidazoles using microwave (MW) heating in both procedures. Combination of two green techniques is reported to give better results in terms of short reaction time and high yield of products. Catalytic efficiencies of both the ionic liquid and heteropolyacid are well documented. The unique thermodynamic properties of both are increasingly enticing chemists to explore their use as media as well as promoters in organic synthesis [31-33], however, high cost of the often used imidazole based ILs and the heteropoly acids deter their use in industrial processes. Herein, we explored the possibility of using simple and cost effective secondary amine based IL namely di-n-propylammonium hydrogensulphate in the synthesis of imidazoles in a solvent free multicomponent reaction (MCR) using MW heating. In a related study, the efficiency of this IL vis a vis a heteroploy acid as an alternative catalyst for the synthesis was examined under identical experimental conditions. Initially the IL and the HPA used were prepared by adopting a simple atom economy procedure using cheap and easily available substrates. The IL used herein was prepared by the action of conc. H2SO4 on di-n-propylamine [34] and the lacunary and defective Keggin type HPA namely H6PAlMo11O40 was prepared by a reported procedure [35] albeit with minor modification. Microwave heating was used while preparing the HPA instead of conventional heating which resulted in less reaction time and high yield of the HPA.
For the purpose of optimization of the amount of catalyst and the reaction time, representative reaction using benzil, benzaldehyde, benzylamine and NH4OAc as the substrates was performed by varying the concentration of the catalyst from 5 mol% to 10 mol% and concomitant variation of reaction time from 2 mins to 10 mins. In case of IL as the catalyst, yield of the desired product 4a improved by 20% when mol% was increased from 5% to 10% and in case of HPA 5 mol% of catalyst and 10 min reaction time gave the best results. No improvement in yield was apparent with increase in mol% of catalyst and/or the reaction time. Results summarized in Table 1 indicated best results with 10 mol% of IL as catalysts and in case of HPA it was 5 mol%.
These two catalysts were used in turn for performing a one-pot four component reaction involving 1,2-diketone, aromatic aldehyde, 1˚-amine and NH4OAc to give the target 1,2,4,5-tetrasubstituted imidazole under solvent free condition mediated by MW. In a typical procedure, the reactants and the catalyst in the form of either HPA or IL, are thoroughly mixed and exposed to MW irradiation. In order to establish the generality of the synthetic procedure, several diversified examples were studied by varying the 1˚-amine and the aldehyde. The yields obtained by using both HPA and the IL are good to excellent with very little or no side products which is a significant improved over other reported procedures. Further good results have been obtained with both the aliphatic as well as the aromatic amines. Reactions with the IL as promoter required 2 - 3 min for completion and reactions catalyzed by HPA required 10 - 13 min. These results are indicative of better efficiency of IL over HPA in this synthesis. The reaction is shown in Scheme 1 and the results summarized in Table 2. Work up was accomplished by extraction of the reaction mixture with CH2Cl2 to separate out the target product. The CH2Cl2 solution was washed with water (3x 50 mL) to remove any trace of the impurity. Since both the catalysts are insoluble in CH2Cl2 the catalysts were precipitated out after addition of CH2Cl2 to the reaction mixture and recovered by simple filtration. The recovered IL was stored in desiccator for reuse and HPA was dried at 85˚C for 24 hours and reused. Both the catalysts retained their activity after two subsequent reuses.
In order to preclude other possibilities, the reaction was examined using related catalyst namely tetra-npropylammonium bromide and NaHSO4 under MW irradiation. With tetra-n-propylammonium bromide, the reaction did not proceed at all whereas with NaHSO4 about
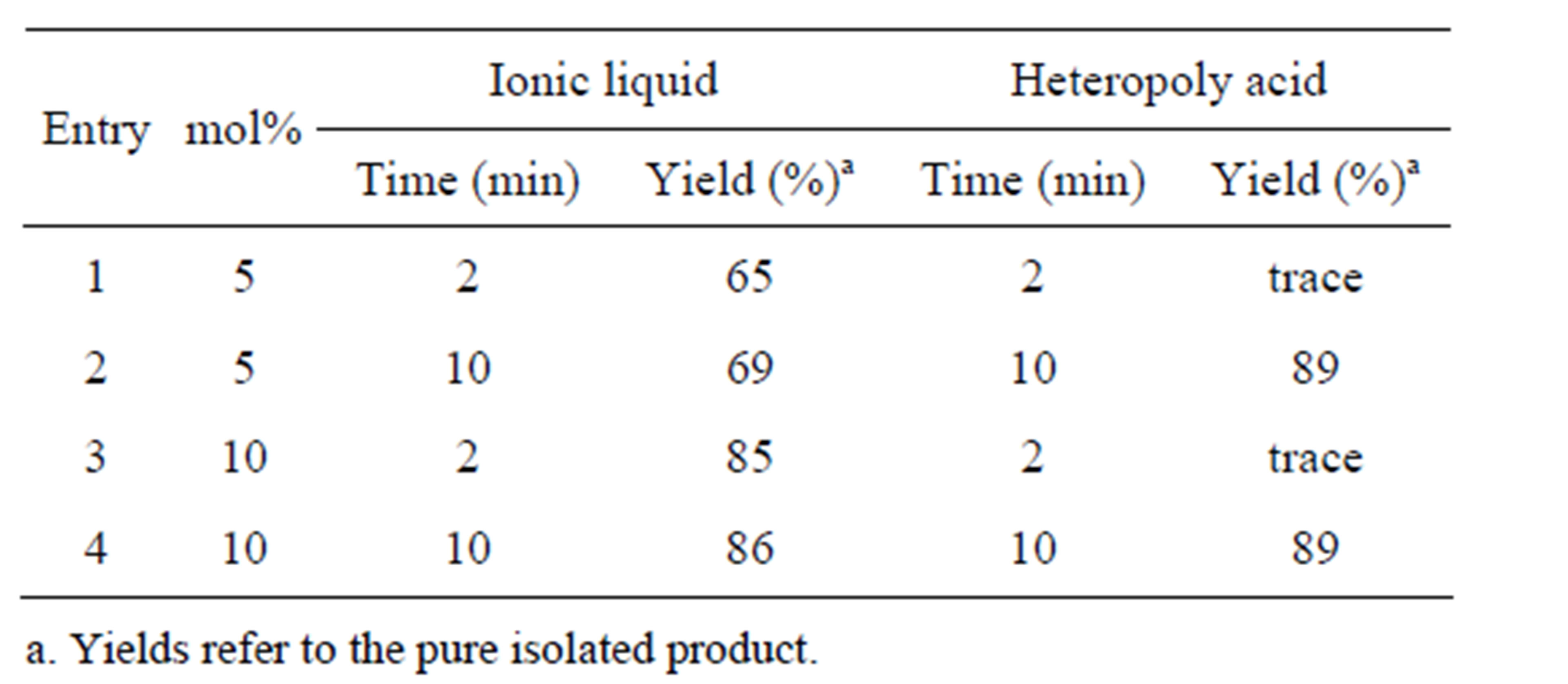
Table 1. Effect of amount of catalyst for the formation of 4a via the 4-MCR of 1, 2, 3 and NH4OAc.
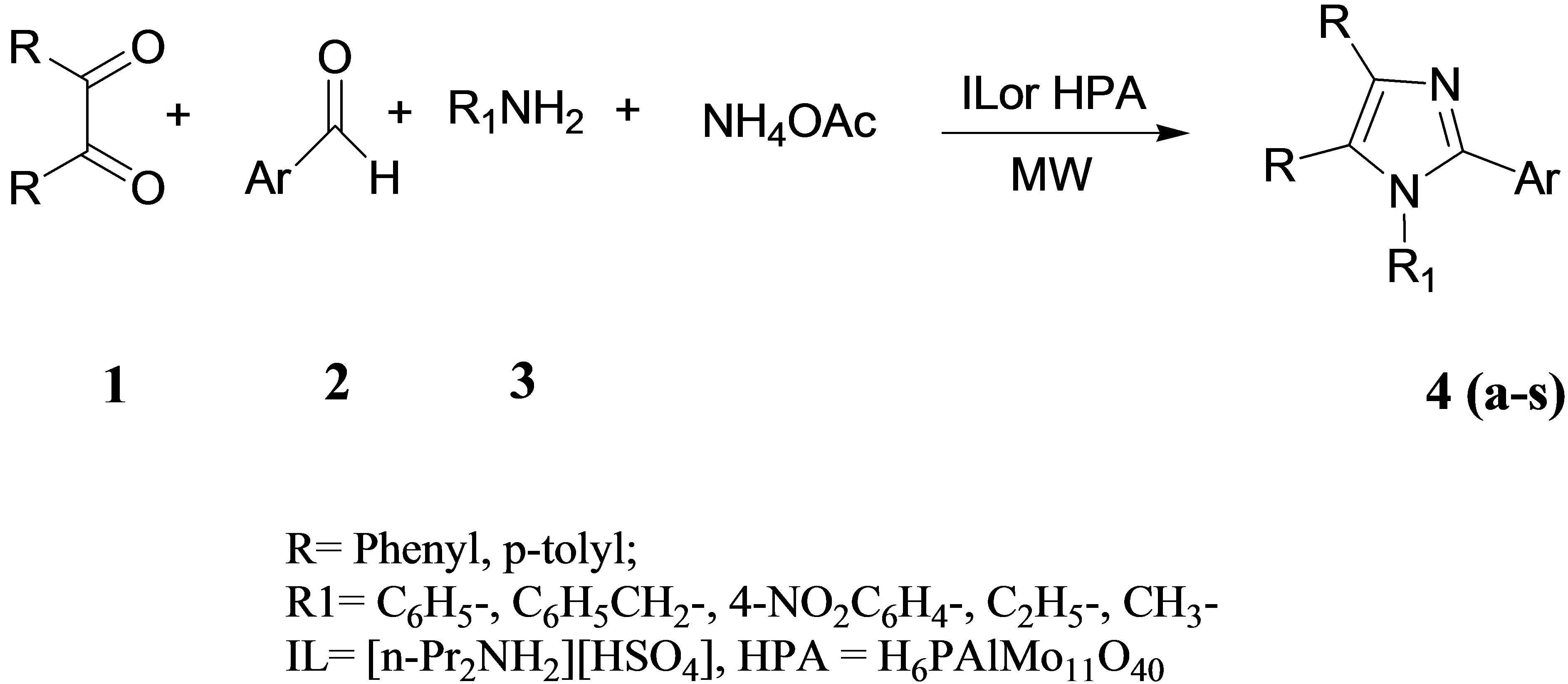
Scheme 1 . Synthesis of imidazole derivatives mediated by IL/HPA.
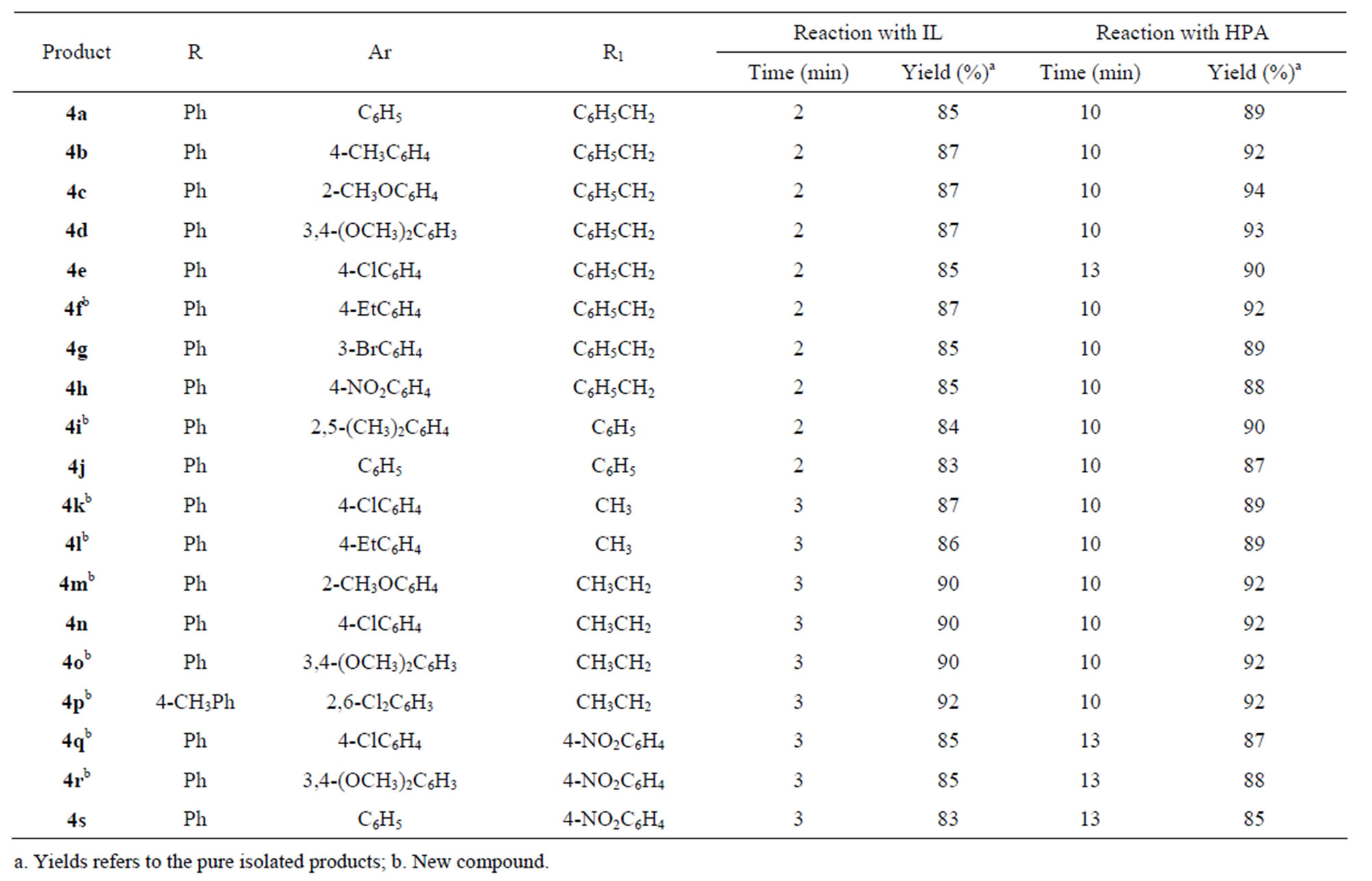
Table 2. Solvent free MW induced one-pot synthesis of 1, 2, 4, 5-tetrasubstituted imidazole promoted by [n-Pr2NH2][HSO4] or HPA using ammonium acetate as nitrogen source.
40% yield of desired product was obtained indicating the essentiality of di-n-propylammonium hydrogensulphate as the appropriate catalyst for the reaction. The results obtained with HPA, [n-Pr2NH2][HSO4], NaHSO4, nPr4NBr and in absence of catalyst for a model reaction are summarized in Table 3. In this study benzil, 2-anisaldehyde, benzyl amine and ammonium acetate were taken as the reference reactants and 10 mol% amounts of catalysts were used.
Investigations were also carried out using urea as the source of nitrogen in the reaction. It was observed that the reaction proceeded equally well with urea in the presence of both IL and HPA. Here efficiency was examined in some model reactions in presence of either IL or HPA. The results obtained by using urea instead of NH4OAc are summarized in Table 4.
All products obtained were characterized by spectroscopic method such as IR, 1H NMR, 13C NMR, Mass spectrometry and by comparing their melting points with those reported in literature. Some new compounds are also reported. Single crystal X-ray analysis of one of the new products namely 1-ethyl-2-(2’, 6’-dichlorophenyl)-4, 5-di(4’-methylphenyl)-imidazole (4p) confirms the structure (Figure 1). Crystals were obtained from ethanol in which case the results indicate the presence of residual
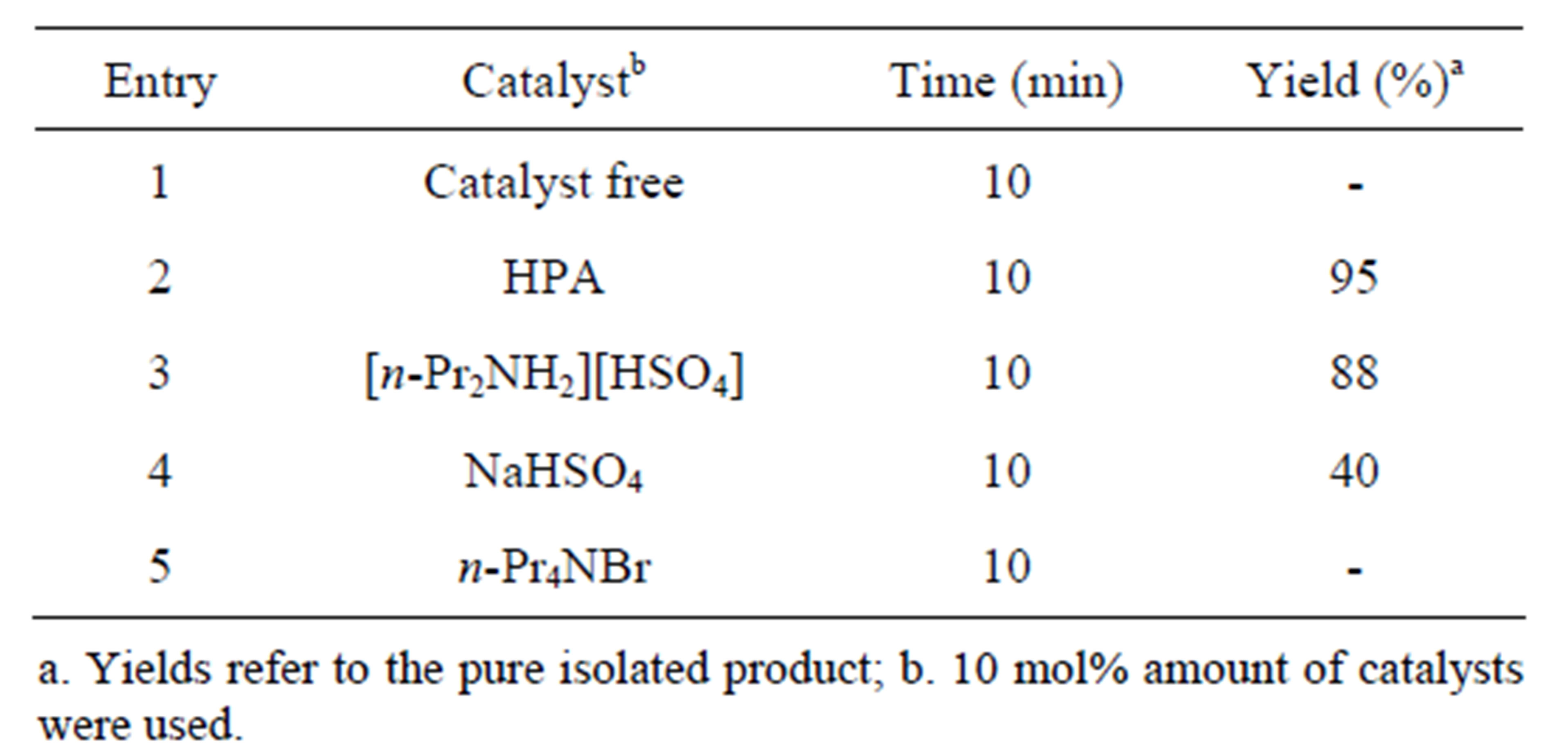
Table 3. Effect of catalyst on the one-pot four component synthesis of tetra substituted imidazole 4c with the usual reactants and NH4OAc.
solvent in the crystal. Attempt to remove the solvent destroyed crystallinity of the product and no worthwhile data could be obtained. Crystallographic data are given in Table 5. CCDC reference number for the compound is CCDC 892443.
3. Conclusion
In conclusion, a one-pot solvent free MCR promoted by the combined use of microwave and IL or HPA offer easy access to tetrasubstituted imidazoles in excellent yield. Procedural simplicity, short reaction time, solvent
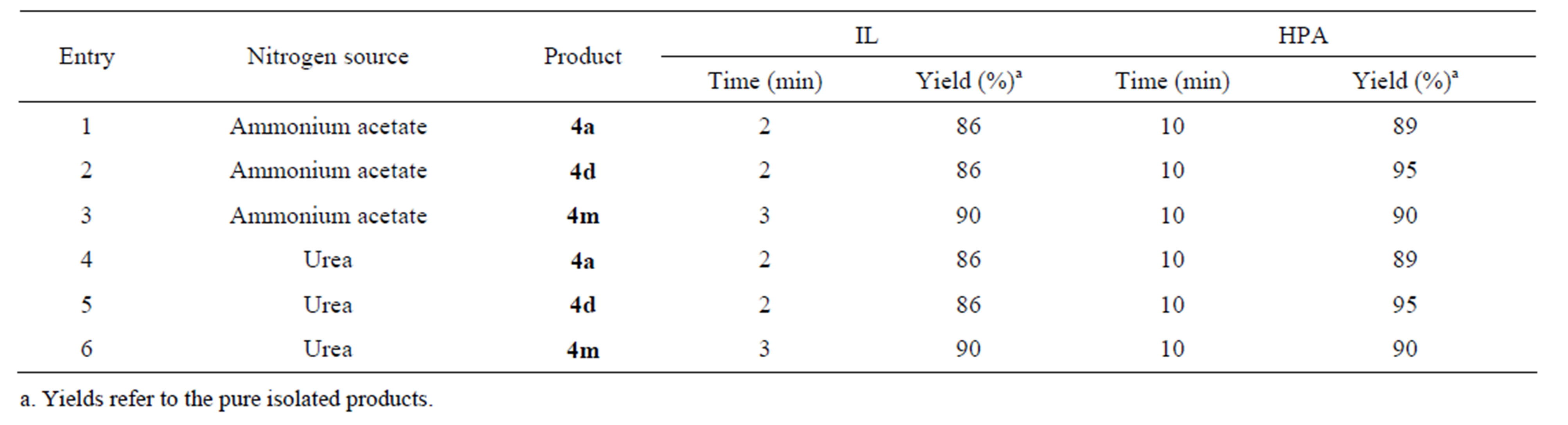
Table 4. The effect of ammonium acetate and urea as the nitrogen source on the 4-MCR to form 4a, 4d and 4m.

Table 5. Crystallographic parameter of compound 4p.
free condition and the use of non toxic and environmentally benign promoters are some of the important features of this new methodology. An added advantage is the simple recovery and efficient reusability of the catalyst. The reaction proceeds equally well with urea as a source of nitrogen instead of NH4OAc which result in cost reduction of the transformation.
4. Experimental Section
4.1. General
Melting points were recorded in a VMP-D model melting point apparatus and are uncorrected. 1H NMR and 13C NMR spectra were recorded on a Bruker advance digital 300 MHz spectrometer in CDCl3. TMS was used as in-
ternal standard. IR spectra were obtained on a Perkin Elmer FT-IR 1600 spectrophotometer using KBr pallets. Mass spectra were recorded on a Waters Micromass ZQTM 400 Mass spectrometer system. The XRF analysis was carried out in Axios XRF Spectrometer. Microwave irradiation of the reaction mixture was performed in a reactor procured from CatalystTM (India) reactor. The single crystal X-ray diffraction data for compound 1- ethyl-2-(2’, 6’-dichlorophenyl)-4, 5-di(4’-methylphenyl) imidazole (4p) was collected on a SMART Bruker ApexII diffractometer. The structure was solved by direct method and refined by full-matrix least squares based on F2 using the SHELXTL 5.1 software package. The hydrogen atoms residing in the carbon atoms were located geometrically. All non-hydrogen atoms were refined anisotropically.
4.2. Synthetic Procedure for the Preparation of the H6PAlMo11O40
A stoichiometric mixture of H3PO4 85% (0.58 g, 0.01 mol), Al2O3∙6H2O (1.21 g, 0.005 mol) and MoO3 (14.4 g, 0.10 mol) was suspended in 150 ml of distilled water and irradiated with microwave for 10 minutes (560 W). On cooling the mixture to room temperature the insoluble MoO3 precipitated and recovered by filtered. Reduced pressure removal of water and drying at 85˚C for 24 hours gave dark green crystals of composition H6PAlMo11O40 identical to that obtained earlier [35]. The metal composition was established by XRF analysis. IR (KBR): ν 1074, 982, 875, 765, 362 and 345 cm−1.
4.3. General Procedure for the Synthesis of Tetrasubstituted Imidazole
A mixture of 1, 2-diketone (1 mmol), aldehydes (1 mmol), primary amines (1 mmol), ammonium acetate (or urea) (1.5 mmol) and IL (10 mol%)/HPA (5 mol%) was irradiated with microwave (560 W) with stirring for 1 - 3/10 - 13 min as mentioned in Table 2. On completion of the reaction monitored by TLC, the reaction mixture was extracted with CH2Cl2 (5 mL × 3), washed with water, dried with anhydrous Na2SO4 and solvent removed under reduced pressure. The crude products were purified by recrystallization from ethanol.
1-Benzyl-2,4,5-triphenylimidazole (4a): Mp 159 - 161˚C (EtOH); IR(KBr): ν 2890 (CH), 1590 (CN), 1480 cm−1; 1H NMR (300 MHz, CDCl3): δH ppm 7.58 - 6.82 (m, 20H, Ar), 5.12 (s, 2H, CH2); 13C NMR (75 MHz, CDCl3): δ ppm 148.08, 138.07, 137.56, 134.46, 131.08, 131.02, 130.94, 130.05, 129.08, 128.92, 128.81, 128.61, 128.59, 128.09, 127.36, 126.78, 126.37, 126.02, 48.28.
1-Benzyl-2-(4’-methylphenyl)-4,5-diphenylimidazole
(4b): Mp 164 - 166˚C (EtOH); IR(KBr): ν 2885 (CH), 1580 (CN), 1482 cm−1; 1H NMR (300 MHz, CDCl3): δH ppm 7.58 - 6.81 (m, 19H, Ar), 5.11 (s, 2H, CH2), 2.38 (s, 3H, CH3); 13C NMR (75 MHz, CDCl3): δ ppm 148.21, 138.84, 137.67, 134.52, 131.07, 129.28, 128.94, 128.75, 128.55, 128.06, 126.78, 125.99, 48.24, 21.36.
1-Benzyl-2-(2’-methoxyphenyl)-4,5-diphenylimidazole
(4c): Mp 188 - 190˚C (EtOH); IR(KBr): ν 2823 (CH), 1593 (CN), 1460 (CC), 1250 cm−1; 1H NMR (300 MHz, CDCl3): δH ppm 7.59 - 6.82 (m, 19H, Ar), 5.09 (s, 2H, CH2), 3.83 (s, 3H, OCH3); 13C NMR (75 MHz, CDCl3): δ ppm 160.04, 147.94, 137.61, 131.02, 130.37, 128.72, 128.53, 128.02, 127.27, 126.72, 126.25, 125.92, 123.29, 113.95, 55.27, 48.16; Anal. Calcd for C29H24N2O: C, 83.63; H, 5.81; N, 6.73, O, 3.84. Found: C, 83.65; H, 5.83; N, 6.72; O, 3.84; HRMS (ESI): m/z = 416.1889 ([M]+), calcd for C29H24N2O: 416.1889.
1-Benzyl-2-(3’,4’-dimethoxyphenyl)-4,5-diphenylimidazole (4d): Mp 163 - 165˚C (EtOH); IR(KBr): ν 2835 (CH), 1600 (CN) cm−1; 1H NMR (300 MHz, CDCl3): δH ppm 7.64 - 6.89 (m, 18H, Ar), 5.10 (s, 2H, CH2), 3.89 (s, 3H, OCH3), 3.69 (s, 3H, OCH3); 13C NMR (75 MHz, CDCl3): δ ppm 149.63, 148.80, 137.99, 134.52, 131.10, 128.89, 128.75, 128.16, 127.42, 126.90, 126.42, 125.96, 121.65, 112.24, 110.99, 55.96, 55.69, 48.28.
1-Benzyl-2-(4’-chlorophenyl)-4,5-diphenylimidazole
(4e): Mp 163 - 165˚C (EtOH); IR(KBr): ν 2832 (CH), 1598 (CN) cm−1; 1H NMR (300 MHz, CDCl3): δH ppm 8.00 - 6.82 (m, 19H, Ar), 5.09 (s, 2H, CH2); 13C NMR (75 MHz, CDCl3): δ ppm 146.92, 137.37, 135.06, 131.08, 130.31, 129.98, 129.11, 128.92, 128.79, 128.20, 126.85, 125.92, 48.34.
1-Benzyl-2-(4’-ethylphenyl)-4,5-diphenylimidazole (4f): Mp 138 - 140˚C (EtOH); IR(KBr): ν 2958 (CH), 1597 (CN), 1492 (CC), 1446, 1388 cm−1; 1H NMR (300 MHz, CDCl3): δH ppm 7.59 - 6.83 (m, 19H, Ar), 5.11 (s, 2H, CH2), 2.67 (q, 2H, J = 6 Hz, CH2), 1.24 (t, 3H, J = 6 Hz, CH3); 13C NMR (75 MHz, CDCl3): δ ppm 148.20, 145.13, 137.88, 137.63, 131.04, 128.97, 128.70, 128.51, 128.08, 128.02, 126.73, 126.26, 125.98, 48.22, 28.66, 15.41. Anal. Calcd for C30H26N2: C, 86.92; H, 6.32; N, 6.76. Found: C, 86.90; H, 6.33; N, 6.78. HRMS (ESI): m/z = 414.2098 ([M]+), calcd for C30H26N2: 414.2096. 1-Benzyl-2-(3’-bromophenyl)-4,5-diphenylimidazole (4g): Mp 148 - 150˚C (EtOH); IR(KBr): ν 2949 (CH), 1601(CN) cm−1; 1H NMR (300 MHz, CDCl3): δH ppm 7.66 - 6.82 (m, 19H, Ar), 5.11 (s, 2H, CH2); 13C NMR (75 MHz, CDCl3): δ ppm 132.17, 131.86, 130.99, 130.00,129.04, 128.86, 128.77, 128.69, 128.57, 128.11, 127.51, 127.22, 126.75, 126.51, 125.98, 125.90, 48.31.
1-Benzyl-2-(4’-nitrophenyl)-4,5-diphenylimidazole
(4h): Mp 170 - 172˚C (EtOH); IR(KBr): ν 2963 (CH), 1608 (CN) cm−1 ; 1H NMR (300MHz, CDCl3): δH ppm 8.12 - 6.73 (m, 19H, Ar), 4.88 (s, 2H, CH2); 13C NMR (CDCl3): δ ppm 148.92, 143.00, 136.57, 133.23, 131.10, 128.93, 128.79, 128.38, 128.05, 126.64, 126.48, 126.41, 124.65, 48.27.
2-(2’,5’-Dimethylphenyl)-1,4,5-triphenylimidazole (4i): Mp 173 - 175˚C (EtOH); IR(KBr): ν 2942 (CH), 1598 (CN), 1492 (CC) cm−1; 1H NMR (300 MHz, CDCl3): δH ppm 7.61 - 6.87 (m, 18H, ArH), 2.23 (s, 3H, CH3), 2.10 (s, 3H, CH3); 13C NMR (75 MHz, CDCl3): δ ppm 147.59, 137.64, 136.50, 134.74, 134.64, 134.51, 131.79, 130.94, 130.78, 130.40, 129.83, 129.73, 129.07, 128.46, 128.37, 128.05, 127.78, 127.71, 127.49, 127.41, 126.44, 20.73, 19.67; Anal. Calcd for C29H24N2: C, 86.97; H, 6.04; N, 6.99. Found: C, 86.90; H, 6.04; N, 6.89. HRMS(ESI): m/z = 400.1939 ([M]+), calcd for C29H24N2: 400.1939.
1-(4’-nitrophenyl)-2-(4’-chlorophenyl)-4,5-diphenylimidazole (4q): Mp 122 - 124˚C (EtOH); IR(KBr): ν 2928 (CH), 1590 (CN), 1489 (CC), 1351 (C-NO2), 739 (CCl) cm−1; 1H NMR (300 MHz, CDCl3): δH ppm 8.05 - 7.03 (18H, m, ArH); 13C NMR (75 MHz, CDCl3): δ ppm 194.66, 152.50, 134.94, 132.87, 129.88, 129.02, 128.60, 127.77, 127.54, 126.47, 126.33, 113.31; Anal. Calcd for C27H18N3O2Cl: C, 71.76; H, 4.01; N, 9.30; O, 7.08; Cl, 7.85. Found: C, 71.68; H, 4.04; N, 9.32; O, 7.09; Cl, 7.83. HRMS (ESI): m/z = 451.1089 ([M]+), calcd for C27H18N3O2Cl: 451.1088.
1-(4’-Nitrophenyl)-2-(3’,4’-dimethoxyphenyl)-4,5-diphenylimidazole (4r): Mp 125 - 126˚C (EtOH); IR(KBr): ν 2939 (CH), 1597 (CN), 1489 (CC), 1346 (C-NO2), 1246 (CO) cm−1; 1H NMR (300 MHz, CDCl3): δH ppm 8.14 - 8.11 (m, 2H, ArH), 7.59 - 7.56 (m, 2H, ArH), 7.30 - 7.12 (m, 12H, ArH), 6.70 (s, 1H, ArH), 3.86 (s, 3H, OCH3), 3.78 (s, 3H, OCH3); 13C NMR (75 MHz, CDCl3): δ ppm 149.60, 133.72, 131.02, 129.90, 129.18, 128.78, 128.56, 128.24, 127.40, 126.99, 124.36, 121.91, 112.28, 110.63, 55.83; Anal. Calcd for C29H23N3O4: C, 72.94; H, 4.85; N, 8.80; O, 13.40. Found: C, 72.96; H, 4.83; N, 8.82; O, 13.49. HRMS (ESI): m/z = 477.1689 ([M]+), calcd for C29H23N3O4: 477.1689.
4.4. Synthesis of Compounds 4(k-p)
Carried as per the general procedure with 0.5 mL of aliphatic amines (methyl amine and ethyl amine).
1-methyl-2-(4’-chlororphenyl)-4,5-diphenylimidazole
(4k): Mp 192 - 194˚C (EtOH); IR(KBr): ν 2925 (CH), 1604 (CN), 1483 (CC), 735 (CCl) cm−1; 1H NMR (300 MHz, CDCl3): δH ppm 7.71 - 7.18 (m, 14H, ArH), 3.50 (s, 3H, NCH3); 13C NMR (75 MHz, CDCl3): δ ppm 146.79, 137.98, 134.90, 134.44,130.97, 130.89, 130.84, 130.32, 129.38, 129.15, 129.05, 128.92, 128.77, 128.20, 127.01, 126.54, 33.29; Anal. Calcd for C22H17N2Cl: C, 76.63; H, 4.97; N, 8.12; Cl, 10.28. Found: C, 76.65; H, 4.99; N, 8.19; Cl, 10.31; HRMS(ESI): m/z = 344.1082 ([M]+), calcd for C22H17N2Cl: 344.1080.
1-methyl-2-(4’-ethylphenyl)-4, 5-diphenylimidazole (4l): Mp 127 - 130˚C (EtOH); IR(KBr): ν 2920 (CH), 1597 (CN), 1480 (CC) cm−1; 1H NMR(300 MHz,CDCl3): δH ppm 7.67 - 7.19 (m, 14H, ArH), 3.51 (s, 3H, NCH3), 2.73 (q, 2H, J = 6 Hz, CH2), 1.29 (t, 3H, J = 6 Hz, CH3); 13C NMR (75 MHz, CDCl3): δ ppm 134.84, 130.79, 129.84, 128.95, 128.43, 128.00, 127.97, 126.87, 33.06, 28.65, 15.40; Anal. Calcd for C24H22N2: C, 85.17; H, 6.55; N, 8.28. Found: C, 85.19; H, 6.58; N, 8.29. HRMS(ESI): m/z = 338.1786 ([M]+), calcd for C24H22N2: 338.1783.
1-Ethyl-2-(2’-methoxyphenyl)-4,5-diphenylimidazole
(4m):Mp 123 - 125˚C (EtOH); IR(KBr): ν 2924 (CH), 1600 (CN), 1462 (CC), 1260 (CO) cm−1; 1H NMR (300 MHz, CDCl3): δH ppm 7.57 - 7.09 (14H, m, ArH), 3.85 (3H, s, OCH3), 3.77 ( 2H, q, J = 6 Hz ,CH2), 0.90 (3H, t, J = 6 Hz, CH3); 13C NMR (75 MHz, CDCl3): δ ppm 157.53, 144.69, 137.57, 134.76, 132.66, 131.77, 130.92, 130.81, 128.93, 128.68, 128.40, 127.88, 126.72, 125.91, 120.88, 120.66, 110.85, 55.47, 39.47, 15.85; Anal. Calcd for C24H22N2O: C, 81.33; H, 6.26; N, 7.90; O, 4.51. Found: C, 81.41; H, 6.28; N, 7.95; O, 4.56. HRMS(ESI): m/z = 354.1736 ([M]+), calcd for C24H22N2O: 354.1732.
1-Ethyl-2(4’-chlorophenyl)-4,5-diphenylimidazole
(4n): Mp 311 - 313˚C (EtOH); IR(KBr): ν 2930 (CH), 1596 (CN)cm−1; 1H NMR (300 MHz, CDCl3): δH ppm 7.68 - 7.16 (14H, m, ArH), 3.94 (2H, q, J = 6 Hz, CH2), 1.03 (3H, t, J = 6 Hz, CH3); 13C NMR (75 MHz, CDCl3): δ ppm 194.57, 146.04, 137.95, 134.90, 134.33, 132.91, 131.26, 130.98, 130.34, 129.89, 129.82, 129.66, 129.08, 129.01, 128.87, 128.76, 128.05, 126.70, 126.30, 39.65, 16.22.
1-Ethyl-2-(3’,4’-dimethoxyphenyl)-4,5-diphenylimidazole (4o): Mp 173 - 175˚C (EtOH); IR(KBr): ν 2928 (CH), 1608 (CN), 1482(CC), 1258 (CO) cm−1; 1H NMR (300 MHz, CDCl3): δH ppm 7.54 - 6.98 (13H, m, ArH), 3.96 - 3.90 (8H, m, 2OCH3 and CH2), 1.02 (3H, t, J = 6 Hz, CH3); 13C NMR (75 MHz, CDCl3): δ ppm 149.64, 149.04, 137.54, 134.63, 131.66, 131.08, 129.21, 129.08, 128.66, 128.04, 126.77, 126.18, 124.11, 121.51, 112.72, 110.98, 56.04, 55.99, 39.65, 16.28; Anal. Calcd for C25H24N2O2: C, 78.10; H, 6.29; N, 7.29; O, 8.32. Found: C, 78.14; H, 6.27; N, 7.32; O, 8.35. HRMS (ESI): m/z = 384.1839 ([M]+), calcd for C24H22N2O: 384.1838.
1-Ethyl-2-(2’,6’-dichlorophenyl)-4,5-di(4’-methylphenyl)imidazole (4p): Mp 135 - 137˚C (EtOH); IR(KBr): ν 2924 (CH), 1605 (CN), 1450(CC), 738 (CCl) cm−1; 1H NMR (300 MHz, CDCl3): δH ppm 7.46 - 7.00 (11H, m, ArH), 3.66 (2H, q, J = 6 Hz, NCH2), 2.45 (3H, s, CH3), 2.28 (3H, s, CH3), 1.00 (3H, t, J = 6 Hz, CH3); 13C NMR (75 MHz, CDCl3): δ ppm 141.10, 138.43, 137.28, 135.59, 131.78, 131.22, 130.86, 129.72, 128.65, 128.22, 128.08, 126.54, 39.32, 21.40, 21.10, 15.88; Anal. Calcd for C25H22N2Cl2: C, 71.26; H, 5.26; N, 6.65; Cl, 16.83. Found: C, 71.29; H, 5.32; N, 6.64; Cl, 16.84. HRMS (ESI): m/z = 420.1160 ([M]+), calcd for C25H22N2Cl2: 420.1160.
5. Acknowledgements
J. D. is grateful to CSIR, New Delhi for award of SRF. The authors are also grateful to SAIF, Gauhati University for recording the single crystal XRD data and Mr S.J. Bharali for analyzing the data.
NOTES