Motor and molecular analysis to detect the early symptoms in a mouse amyotrophic lateral sclerosis model ()
1. INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a fatal progressive neuromuscular disorder, with most patients dying within 3 to 5 years from diagnosis. Although most cases of ALS are sporadic in origin, 5% - 10% are familial (FALS). Among the population with FALS, 10% possess a mutation in the gene that codes for the enzyme Cu/Zn superoxide dismutase (SOD1) [1,2], an ubiquitously ex- pressed protein that plays a key role in oxygen free radical scavenging, catalyzing the dismutation of superoxide 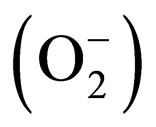 to hydrogen peroxide [3,4]. Recently, other mutations involved in FALS cases have been discovered (TARDBP, FUS, C9ORG72,) [5]. Moreover, the finding of C9orf72 hexanucleotide repeat expansion in both sporadic and familial ALS suggests that many apparently sporadic cases are in fact underestimated familial cases [6,7].
to hydrogen peroxide [3,4]. Recently, other mutations involved in FALS cases have been discovered (TARDBP, FUS, C9ORG72,) [5]. Moreover, the finding of C9orf72 hexanucleotide repeat expansion in both sporadic and familial ALS suggests that many apparently sporadic cases are in fact underestimated familial cases [6,7].
To investigate the pathogenic mechanisms leading to ALS, a number of animal models have been created based on genes involved in ALS pathogenesis [8]. However, the most widely used ALS experimental model remains the SOD1 mouse, even though it does not fully represent the human disease, since only few FALS cases are ascribed to its mutation, but better than others reproducing histopathological and clinical features of the disease. The SOD1 mouse was created by Gurney and colleagues in 1994 [9] by inserting a dominant “gain-of-function” mutation in human SOD1 over-expressed in the transgenic mice (the largest amount of mutant SOD is expressed in the brain). The mutation consists in the amino acid substitution at position 93 from glycine to alanine (G93A) that has little effect on enzyme activity.
The phenotype depends on the number of transgene copies [10,11]. Paralysis (in one or more limbs) is due to progressive loss of motoneurons. The mechanism by which the hSOD1-G93A mutation exerts a toxic effect is poorly understood. Multiple mechanisms have been proposed as causes of motoneuron degeneration including glutamate excitotoxicity [12], protein aggregation [13,14], increased oxidative stress, proteosome dysfunction [15,16] and axonal transport defects [17,18]. The end-stage pattern of disease in these animals is the one most closely mimicking the changes that are generally observed in ASL patients, with filamentous inclusions in both neurons and axonal processes [19].
In order to evaluate the efficacy of any treatment, either pharmacological or cellular, it is very important to treat all animals at the effective early onset of the disease, at the same stage of progression. The outcome of treatment administered at a fixed age-point could result in misleading because of the variability of the onset (1 to 2 weeks). To this aim, we performed a set of three behavioral assessments (neurological condition, Rotarod and Paw Grip endurance tests) to evaluate motor performance that best correlates with the beginning of the symptomatic phase of the disease. Moreover, to explain different interindividual variance in the disease onset, we performed molecular analysis to measure hSOD1 mRNA expression levels in SOD1-G93A mice.
2. MATERIALS AND METHODS
2.1. Animal Care and Use
Male transgenic mice carrying the human mutated superoxide dismutase 1 gene (B6SJL-TgN[SOD1G93A]1 Gur, Jackson Laboratories; stock number 002726) were used; these mice have high transgene copy number, as reported in the datasheet. Founders were kindly gifted by M. Bentivoglio and R. Mariotti (University of Verona). The colony was derived by breeding male transgenic mice to naive (B6xSJL/J)F1 females (Janvier SAS, Le GenestSaint-Isle, France).
All experimental procedures on live animals were performed according to the European Communities Council Directive of 24 November 1986 (86/609/EEC) and University of Torino’s institutional guidelines on animal welfare (DL 116/92). All efforts were made to minimize the number of animals used and their suffering. After completion of the experimental procedure mice were overdosed with pentobarbital (i.p.) and euthanized by cervical dislocation.
2.2. Genotyping Mice
DNA from mouse tail was extracted incubating a small piece (0.5 cm) of tail in 100 ml of lysis buffer (10 mM Tris HCl, 50 mM KCl, 0.01% gelatin, 0.45% IGEPAL, 0.4% Tween-20) and 25 mg of proteinase k at 55˚C over night under gentle shaking. On the extracted DNA we performed quantitative real-time PCR with a SYBR Green core reagent kit in a 7000 real time PCR system (Applied Biosystem) following the manufacturer’s instructions in order to evaluate the presence and the copy number of the human transgene superoxide dismutase-1 (hSOD1). The primers used were: 5’-CATCAGCCCTAATCCATCTGA-3’ and 5’-CGCGACTAACAATCAAAGTGA-3’ for hSOD1 gene; 5’-CTAGGCCACAGAATTGAAAGATCT-3’ and 5’-GTAGGTGGAAATTCTAGCATCATCC-3’ for mouse interleukin 2 gene (mIL2), used as housekeeping gene [20]. Samples were amplified simultaneously in triplicate in 1 assay run. Changes in transgene copy number were determined as the difference in threshold cycle (DCt) between the transgene and the reference gene (Ct hSOD1 - Ct mIL2).
2.3. Evaluation of hSOD1 mRNA Expression
Total RNA from 2-month-old mouse ears was extracted with Trizol following supplier’s instructions (Invitrogen). The first-strand cDNA was synthesized with 10 mg of total RNA using the High Capacity cDNA Reverse Transcription Kit following supplier’s instruction (Applied Biosystems). Quantitative real-time PCR was performed with SYBR green core reagent kit (Applied Biosystems). The primers used were 5’-AAGCATTAAAGGACTGACTGAAGG-3’ and 5’-CACCGTGTTTTCTGGATAGAGG-3’ to measure hSOD1 mRNA level; 5’-TGCACCACCAACTGCTTAGC-3’ and 5’-GGCATGGACTGTGGTCATGAG-3’ to measure glyceraldehyde 3-phosphate dehydrogenase (GADPH) mRNA, used as housekeeping gene for data normalization [21]. Samples were amplified simultaneously in triplicate in 1 assay run. Changes in mRNA levels were determined as the difference in threshold cycle (DCt) between hSOD1 mRNA and the reference gene (Ct hSOD1mRNA - Ct GAPDH).
2.4. Behavioral Tests
We performed the following behavioral tests: 1) neurological scoring of motor deficits by a trained observer; 2) performance on the Rotarod task; 3) Paw Grip Endurance (PaGE); all of which are commonly used to evaluate SOD1-G93A animals [22,23]. Before starting the behavioral tests, the animals were weighed. Experiments were carried out following similar environmental conditions and at the same hour to reduce circadian influences. Beginning at P42 (postnatal day 42), the animals were assessed weekly, and around P80 (before the appearance of the first symptoms) twice per week. The first 2 weeks of tests were considered as training.
Neurological score: the mice were evaluated for signs of motor deficit with the following 4 point scoring system: 4 points if normal (no sign of motor dysfunction); 3 points if hind limb tremors were evident when suspended by the tail; 2 points if gait abnormalities were present; 1 point for dragging of at least one hind limb; 0 points for inability to right itself within 30 seconds.
Rotarod test: we measured the time animals could remain on the rotating cylinder in a 7650 accelerating model of a Rotarod apparatus (Ugo Basile). Each animal was given three trials. The arbitrary cut-off time was 300 seconds.
Paw Grip Endurance (PaGE) test: the animal was placed on the wire-lid of conventional housing cage. The lid was gently shaken to prompt the mouse to hold onto the grid before it was swiftly turned upside down. Grip score was measured as the length of time that the mouse was able to hang on to the grid. The arbitrary cut-off time was 90 seconds.
3. RESULTS
3.1. Behavioral Outcome
For assessing the disease onset, 24 mice underwent a battery of behavioral tests: performance on the Rotarod, PaGE test, scoring of motor deficits by a neurological scale, and weighing. Two experienced investigators performed all behavioral tests.
The first two weeks of tests (starting around P60) were considered as training for the animals, and during this period mice were tested weekly until P80. Thereafter the tests were performed twice a week, to better monitor the animals and precisely detect the first symptoms.
Neurological test. This test was performed in an openfield in order to observe the mouse walking. The main problem of such test is that tremor phase (score 3) was not always evident: therefore it could happen to register for long score 4 (no motor dysfunctions), and then quickly switch to score 2 (gait abnormalities). However, as shown in the graph, this implies that animals displayed a normal posture until P111-P114 and suddenly worsened to 1.5 score in just one week (Figure 1(a)).
Rotarod test. This test measures motor performance and motor coordination of rodents. As showed in the graph, first training weeks were fundamental to animals for learning the task: in fact over the weeks the performance gradually improved and reached a plateau. The trend was quite stable until P100, when the motor performance started worsening. In about 25 days mice became totally unable to carry out the task (Figure 1(b)).
PaGE test. Animals were evaluated with this test to examine the hind limb resistance. Mice were able to finalize the test even without training, probably because this motor activity results more common than Rotarod for rodents. However, similarly to the Rotarod, PaGE test highlighted a sudden worsening in the motor performance from P100, until they were completely unable to perform the task around P128 (Figure 1(c)).
Body weight. We observed a body weight difference between P90 (when animals are adult and ALS onset is not yet evident, as shown by the other tests) and P128 (late-phase of the disease). The weight showed only a modest decrease (about 6%) in the last month (Figure 1(d)).
3.2. Correlation between SOD1 mRNA and Disease Onset
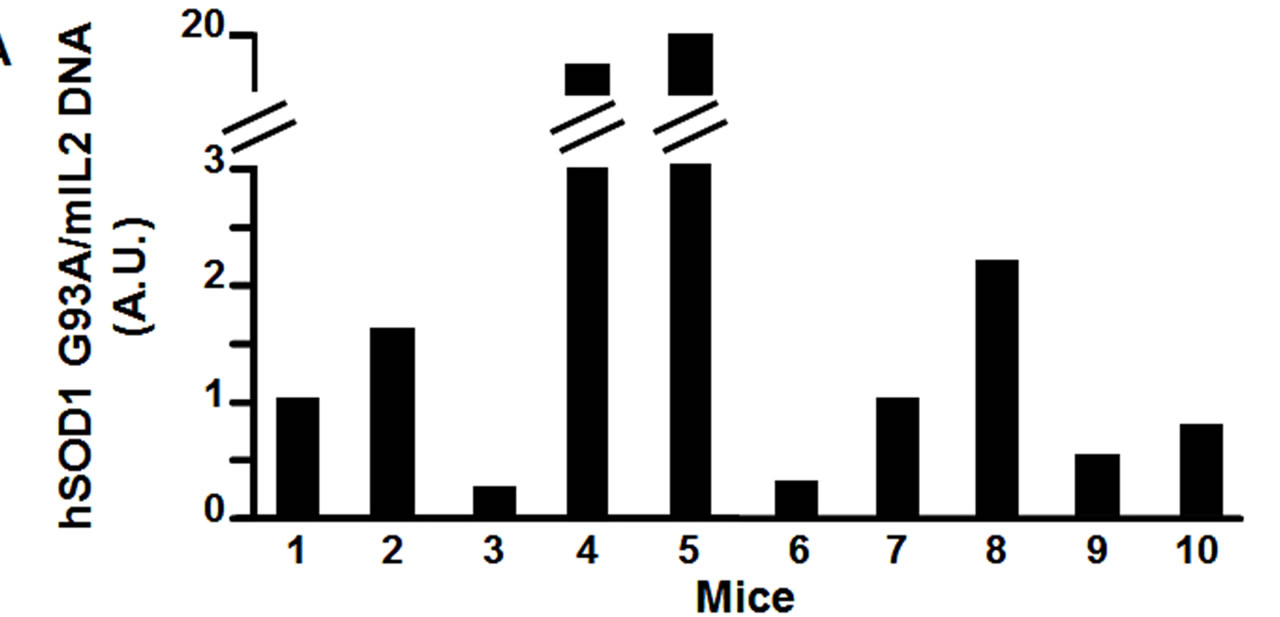
Four animals, out of the 24 analyzed at the behavioural tests, were excluded from the average because they showed the first symptoms of the disease early in advanced compared to the other 20 mice (mice number 1-4, Figure 2). These four animals showed the first symptoms of the disease between P70 and P90, approximately from 20 to 40 days before the average value observed with the other 20 mice (P110).
It has been previously reported that changes in transgene copy number in SOD1-G93A mice can sometimes occur after mating, due to intra locus recombination events, usually resulting in delayed disease onset and an increase in the lifespan of these animals [24].
Therefore, using real time quantitative PCR, we measured the relative transgene copy number in the 4 “atypical” mice, and in 6 mice that showed the first symptoms of the disease approximatively at P110, to investigate the correlation between age of disease onset and transgene copy number (Figure 2(a)). From the ear of the same animals we measured mRNA expression levels of hSOD1 gene at 2 months of age (Figure 2(b)). The ear was chosen because in preliminary experiments (data not shown) we extracted RNA from different tissue samples (ear, spinal cord, cortex, cerebellum, tail, fingers) and we observed a direct correlation in hSOD1 mRNA expression between spinal cord and ear.
 (a)
(a)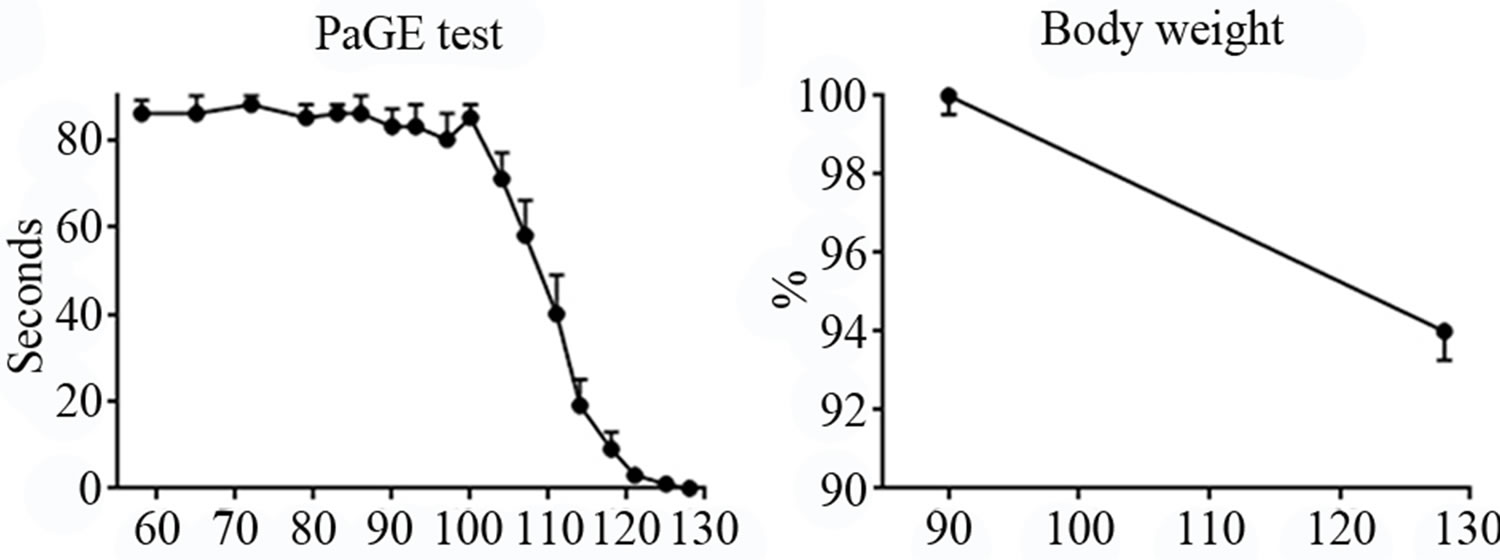 (b)
(b)
Figure 1. Panel A, neurological score of SOD1-G93A mice. Panel B-C, Rotarod and PaGE behavioral tests, respectively. Panel D, weight loss in SOD1-G93A mice.
 (a)
(a)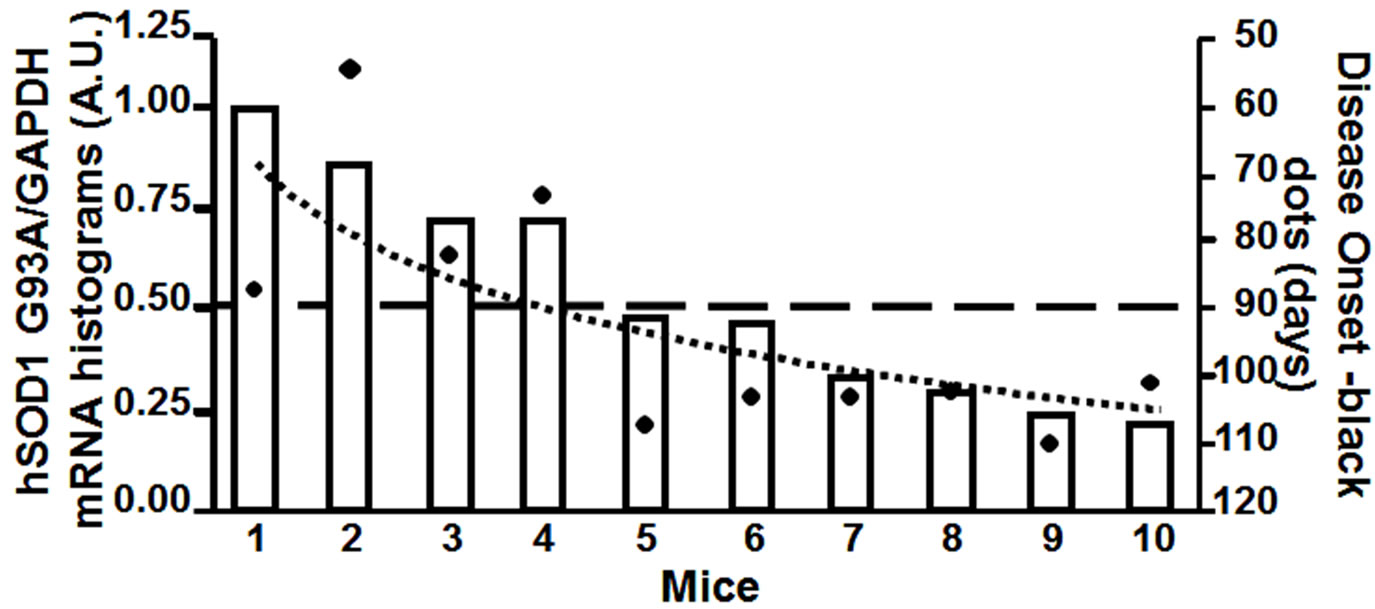 (b)
(b)
Figure 2. Panel A, hSOD1 gene copy number evaluation in SOD1-G93A mice. Panel B, hSOD1 mRNA expression levels in SOD1-G93A mice (histograms) correlated to disease onset expressed in days (black dots).
We arbitrarily considered that, when the mouse showed behavioral deficits in at least two tests (i.e. values < 90 seconds the PaGE test, < 300 seconds in the rotarod test, and score < 4 in the neurological test) for two consecutive measurements, such date corresponded to the disease onset.
Interestingly, our results showed a better correlation between disease onset and mRNA levels than with transgene copy number (Figure 2(b)). In particular, the “atypical” mice number 1, 2, 3 and 4, that showed the first symptoms of the disease earlier than P110, had a hSOD1 mRNA relative level greater than 0.5 (arbitrary unit scale; DCt > −3.5). The remaining mice, with a relative value of hSOD1 mRNA lower than 0.5, approximately around 0.25 (DCt < −2.5), showed the onset of the disease ranging from P100 to P110 as we would expect from this transgenic model. On the other hand, mice number 5, 6, 7 and 8 with quite different transgene copy number (arbitrary unit scale) showed approximately the same age of onset of the disease, around P110. This molecular analysis let us to characterize and to exclude from further studies the animals (mice number 1, 2, 3 and 4) with a too early onset of the disease.
4. DISCUSSION
One of the most widely used animal models of ALS consists in SOD1-G93A mice, which express multiple copies of the mutant form of the human Cu/Zn SOD [24]. However a substantial problem in SOD1-G93A mice is represented by changes in transgene copy number occurring after mating, due to intra locus recombination events: this can result in delayed disease onset and an increase in their lifespan [25]. However, some authors refer to rarely detect the copy number reduction [26].
The expression level of mutated SOD1 can vary independently from the number of copies of the transgene, thus modifying the onset and progression of the disease. In fact, the appearance of the first symptoms varies among the animals, in a time window of 1/2 weeks. Afterwards, the progression of the disease is quite fast, and the animals die approximately in 30 days. Due to the variability of disease onset, preclinical studies should rely on assessment for the precise detection of the first symptoms, instead of starting treatment in each animal at the same pre-defined age-point. Therefore, we used two different approaches, behavioral tests and molecular analysis to evaluate motor functions of male mice, in order to establish the beginning of the disease. We used only male animals, since ALS is characterized by a faster progression in males than in females probably for the partial protective role of estrogens in the latter ones [27]: in previous experiments, we have observed significant differences in terms of survival, motor performance and histological parameters between sexes, according to the literature [23].
In our study, the battery of behavioral tests performed on 24 mice revealed that the Rotarod and the PaGE tests were reliable markers of the onset of the disease.
The assessment of Rotarod performance has widely been used in various models of neurodegenerative diseases to monitor motor coordination, strength and balance: in fact, it is considered one of the most sensitive method for detecting symptoms in hemiplegic and ALSaffected mice [23,28,29].
In our experience, after the first weeks of training, mice can easily perform 300 seconds on the rotating cylinder, until the hind limbs show the first symptoms of weakness [30,31].
Similarly, mice perform very well at the Grip test at the first observation age point (6 weeks of age). They can resist upside down for long, but a cut-off of 90 seconds was set. Additionally we considerably concluded the test even though the mice kept on hanging with the only forelimbs for more than 5 seconds. In this way, we can distinguish between the strength of the forelimbs (involved later in disease) and the hind limbs. Furthermore, the Grip test is less expensive and less time consuming than the Rotarod apparatus, and does not require a training, even though we advise to accustom mice with the test.
Additionally, in contrast with the large number of animals considered, SEM for the Rotarod test was considerably higher than for the PaGE test; therefore, the latter displays a minor variability and a better reliability.
On the other hand, the neurological score evaluation and the body weight loss gave poor results. In particular, only when the disease has been already in an advanced phase (approximately P115 days), we observed a significant decrease in the neurological score and in the weight, according to Knippenger and Coll. (2010) [29] who detected first abnormalities around P109. Moreover, the visual evaluation (neurological score) by an experienced investigator is not sufficient for identifying the first symptoms, since particularly during the first stages, mice appear quite unaltered although the motoneuron degeneration is already ongoing [29].
Additionally, we can not easily explain why sometimes our animals switched straight from a 4 point score (no motor dysfunction) to gait abnormalities (score 2). For all these reasons, we consider that this test is not suitable for an accurate evaluation of the onset of the disease. Nevertheless, for further information, we also report the Weydt’s paper [22] which on the contrary, considers this kind of observation as the most sensitive method for detecting abnormalities.
Similarly, monitoring of the weight loss of the animals is poorly helpful in our opinion as previously reported by Knippenberg [29]. Probably, the decrease becomes lately evident, due to the difficulty in feeding as ALS related-muscular atrophy occurs. Pharmacological treatment of animals at this stage of the disease can unlikely give encouraging results. Therefore, weight and neurological score can only be considered additional parameters to monitor disease progression.
Furthermore, we measured transgene copy number and mRNA expression levels of hSOD1 in 10 out of 24 mice analyzed at the behavioral tests. We considered 4 “atypical” mice that showed the first symptoms of the disease early in advance compared to the other mice, and 6 of the “typical” mice. Interestingly, we observed a better correlation between the onset of the disease and hSOD1 mRNA expression levels in two month-old mice ears, rather than to the transgene copy number.
The inconsistence between hSOD1 mRNA expression and gene copies can be explained by the mislocation of the transgene in a poorly transcribed region. Therefore, the transgene copy number can be a misleading parameter. In fact, even high transgene copy number can produce low mRNA hSOD1 levels in the mouse, corresponding to a late-onset disease (mouse number 5 in Figure 2). By the contrary, mice with low transgene copy number, probably in a highly transcribed region, can develop the disease quite early (mouse number 3 in figure 2). In particular, mice with a relative mRNA level below 0.5 (arbitrary unit) will be considered suitable for further pharmacological experiments whereas mice with higher mRNA levels will be excluded for a too premature disease onset.
For a pharmacological study, it is very important to have a homogenous group of animals (all at the initial phase of the disease). However, sometimes the symptoms onset cannot be arbitrarily fixed; therefore, we employ the behavioral tests for precisely detecting the first symptoms, and we specifically consider this moment (i.e. when the mouse shows at least two behavioral deficits for two consecutive measurements) as the exact disease onset. Simply referring to the animal age could in fact generate unreliable data.
Such approach allows to clearly identify the effects of the treatment on animal behavior and their lifespan: in fact a decrease in the progression of the disease or an increase in the lifespan of the mice of 1 or 2 weeks would represent a very encouraging result, due to short lifespan of this animal.
In conclusion, we show that i) a battery of behavioral tests allows precisely detecting the first symptoms, and the onset of the disease; and ii) there is a direct correlation between hSOD1 mRNA expression and the age of onset of the disease.
The association of behavioral tests and molecular analysis represents a sensible and accurate tool to early detect the murine ALS symptoms and to perform a pharmacological treatment in the best therapeutic window.
5. ACKNOWLEDGEMENTS
This work was supported by Italian Ministry of University and Research (MIUR) grant to A.V.