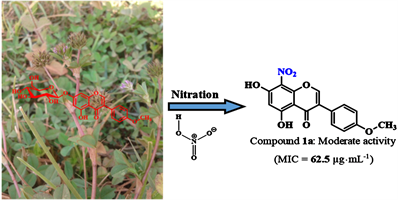
1. Introduction
Antibiotic resistance is one of the biggest threats to global health today [1] . In Cameroon, the work of Mouiche et al. (2019) shows the high prevalence of this phenomenon of resistance to antibiotics commonly used in humans [2] . So, more than ever, it becomes urgent and vital to find new effective drugs especially since it is estimated that if this is not done, there would be no more antibiotics capable to fight against deadly resistant pathogens by 2050 [3] . In this perspective, nitro and acetylated aromatic compounds are a promising avenue for the discovery of new antibiotic substances. Indeed, it has been demonstrated that aromatic compounds with one or more nitro groups directly attached to the aromatic nucleus exhibit pharmacological properties allowing them to be used as antibiotics [4] . Furthermore, it has been shown that the nitro group, present in antibiotic substances, plays a key role due to its physical and chemical properties such as its polarity, electronegativity, small size, redox properties as well as its ability to establish hydrogen bonds [5] . Figure 1 below shows some structures of flavonoid derivatives obtained after the nitration reaction. It is also reported in the literature that the acetyl group increases the antimicrobial activity of certain compounds like flavonoid glycosides [6] . Therefore, in our continued search for new antibiotics compounds [7] , we have undertaken the nitration and acetylation of a naturally occurring isoflavonoid, namely sissotrin previously isolated in our Research Unit from Trifolium baccarinii Chiov (Fabaceae) [8] . In this paper, we report the purification, structure elucidation as well as the evaluation of the antibacterial activity of the semisynthetic derivatives obtained after performing nitration and acetylation reactions.
2. Material and Methods
2.1. Extraction and Isolation of Substrate
Extraction as well as isolation and characterization of sissortrin (1) from Trifolium baccarinii were performed by a method previously described by Tsamo et al. (2021) [8] .
2.2. Chromatography Methods
Column chromatography was carried out on silica gel 230 - 400 mesh, Merck
![]()
Figure 1. Some flavonoids derivatives obtained by nitration.
(Merck, Darmstadt, Germany), 70 - 230 mesh (Merck) (VWR, France). Thin-layer chromatography (TLC) was carried out on Merck pre-coated silica gel (60 F254) aluminium foil (Merck) and the spots were visualized by an UV lamp multiband UV-254/365 nm (Model UVGL-58, Upland, CA 91786, USA) followed by spraying with 50% H2SO4 followed by heating at 100˚C.
2.3. Nuclear Magnetic Resonance (NMR) Analysis
The 1H and 13C-NMR spectra were recorded on a Bruker Avance III 600 spectrometer equipped with a cryoplatform (1H at 500 or 600 MHz and 13C at 125 or 150 MHz). 2D NMR experiments were performed using standard Bruker microprograms (Xwin-NMR version 2.1 software). All chemical shifts (δ) were reported in parts per million (ppm) using the residual solvent signals as secondary reference relatively to TMS (δ = 0) as an internal standard, while the coupling constants (J) were given in hertz (Hz). Deuterated solvents, methanol (CD3OD) and dimethyl sulfoxide (DMSO-d6) were used as solvents for the NMR experiments.
2.4. Spectrometric Analysis
High resolution mass spectra were obtained with QTOF (Quadrupole Time of Flight) Compact Spectrometer (Bruker, Germany) equipped with a HRESI source. The spectrometer was operated in positive and negative modes (mass range: 50 - 1500, with a scan rate of 1.00 Hz) with automatic gain control to provide high-accuracy mass measurements within 0.4 ppm deviation using Na formate as calibrant. The following parameters were used for experiments: spray voltage of 4.5 kV, capillary temperature of 200˚C. Nitrogen was used as sheath gas (4 L/min).
2.5. Semisynthesis and Purifications
2.5.1. Nitration of Sissotrin (1)
The reaction was carried out using the method previously described by Lewin et al. (2010) with slight modifications (Scheme 1) [9] . A mass of 200 mg (448.431 μmol) of sissotrin (1) was dissolved in a 100 mL flask with 40 mL of acetic acid
![]()
Scheme 1. Nitration and acetylation of sissotrin (1).
(CH3COOH). Then, 35 μL of fuming nitric acid (HNO3, 70%) was added to the solution obtained. The mixture was refluxed at 70˚C and the reaction was monitored on a TLC plate. After 2 hours, the reaction medium underwent differential solubilization with n-BuOH (120 mL, H2O; 80 mL, n-BuOH, repeated 3 times). The organic phase obtained was evaporated on a rotary evaporator and under reduced pressure. The purification of the mixture of products obtained on silica gel column chromatography using n-hexane/ethyl acetate (50:50 → 00:100, v/v) as eluent gave: 8-nitrobiochanin A (1a: 7.10 mg; Rf 0.60) and 6-nitrobiochanin A (1b: 8.20 mg; Rf 0.40) (Scheme 1).
8-nitrobiochanin A (1a): Yellow powder from n-hexane/ethyl acetate, (20:80, v/v); positive HRESIMS m/z: 330.0566 [M + H]+ (calcd for C16H12NO7, 330.0608). 1H NMR (CD3OD, 600 MHz) and 13C NMR (CD3OD, 150 MHz) data, see Table 1 and Table 2.
6-nitrobiochanin A (1b): Yellow powder from n-hexane/ethyl acetate, (20:80, v/v); positive HRESIMS m/z: 330.0609 [M + H]+ (calcd for C16H12NO7, 330.0608). 1H NMR (CD3OD, 600 MHz) and 13C NMR (CD3OD, 150 MHz) data, see Table 1 and Table 2.
2.5.2. Acetylation of Sissotrin (1)
In a 50 mL capacity Erlenmeyer flask containing the substrate (1, 100 mg, 2.2 mmol), we added 10 mL of pyridine in order to solubilize and then 1 mL of acetic anhydride. The mixture was stirred at room temperature. After 3 hours, the reaction medium was controlled by means of an analytical TLC which showed that the latter no longer contained any substrate. A little amount of water (90 mL) was added to this reaction medium and the solution obtained was subjected to differential solubilization in a separatory funnel (250 mL) with 100 mL of ethyl acetate until complete exhaustion. The organic phase obtained was purified after drying by column chromatography over silica gel (n-hexane/ethyl acetate 80:20, v/v) to yield to a known derivative identified as 2",3",4",6"-tetraacetylsissotrin (1c: 110.6 mg; Yield 80.34 %; Rf 0.55) (Scheme 1).
![]()
Table 1. 1H NMR data of compounds 1a, 1b, 1c and 1.
aConfused on the spectrum.
![]()
Table 2. 13C NMR data of compounds 1a, 1b, 1c and 1.
2",3",4",6"-tetraacetylsissotrin (1c): Yellowish powder from n-hexane/ethyl acetate, (60:40, v/v); positive ESI-MS: m/z, 637.16 [M + Na]+. 1H NMR (DMSO-d6, 600 MHz) and 13C NMR (DMSO-d6, 150 MHz) data, see Table 1 and Table 2.
2.6. Antibacterial Assay
2.6.1. Bacterial Strains and Sample Preparation
The studied microorganisms consisted of six bacterial strains among which three Gram-positive (Staphylococcus aureus ATCC 43300, Methicillin-resistant Staphylococcus aureus 33591 and Staphylococcus aureus CPC) and three Gram-negative (Escherichia coli ATCC 25922, Shighella flexineri CPC and Shighella flexineri NR 518) all previously stored in 80˚C. Mueller Hinton Agar (MHA) was used to revive the bacterial strains 24 h before the experiment. They were obtained from our local stocks. The substrate as well as the semisynthetic derivatives were weighed and solubilized in dimethyl sulphoxide (DMSO) to obtain a final concentration of 20 µg∙mL−1. Ciprofloxacin (Sigma Aldrich, USA) used as reference drug was prepared in sterile distilled water at 1 µg∙mL−1.
2.6.2. Determination of the Minimum Inhibitory Concentration (MIC)
Minimum Inhibitory Concentrations of the samples were determined according to the microdilution method described by the Clinical and Laboratory Standards Institute in 2012, [10] using 96-well microtiter plates. Briefly, 190 μL of culture medium (Nutrient Broth) was introduced into the first line of the plate and 100 μL in the rest of the plate. Thereafter, 10 μL of test products were added exclusively in the wells of the first line to reach a final volume of 200 μL. After slight homogenization, serial two-fold dilutions of test compounds were made through a transfer of 100 μL of the mixture from the first well to the second and so on to the last. The final concentrations in the wells ranged from 500 - 3.906 µg∙mL−1 for the test compounds and 119 - 0.116 µg∙mL−1 for ciprofloxacin. Then 100 μL of bacteria inoculum standardized to 1.5 × 106 CFU mL−1 were introduced into each well. The sterility control consisted of the culture medium (Nutrient Broth) alone, while the negative control consisted of the medium and bacteria (Nutrient Broth + bacteria). Ciprofloxacin (Sigma-Aldrich, Germany) + bacteria was used as a positive control. The plates were incubated at 37˚C for 24 hours and after this period, the turbidity was observed as an indication of growth. The MIC was considered as the lowest concentration with no visible growth of microorganisms (no turbidity). All tests were performed in triplicate.
3. Results and Discussion
3.1. Chemical Analysis
The nitration reaction of 200 mg of compound 1 led to two products, 1a and 1b, with masses of 7.10 mg and 8.20 mg respectively, while the 100 mg acetylation reaction of the same compound led to a single product, 1c with a mass of 110.6 mg, giving a yield of 80.34%. A study of the literature reveals that the presence of a methoxyl group in 4' promotes nitration on the ring A, in particular in positions 6 and 8 of the 5,7-dihydroxylated flavonoids [11] . However, position 8 being the most favored [12] which was not the case for our reaction. The low yield obtained in the first reaction is justified by the fact that it proceeds in several steps, in particular in three steps (with loss of the osidic part) and it resulted in more than one product as presented in Scheme 1. In contrast, in the second reaction there is no loss of the osidic moiety.
Compound 1b obtained from the nitration reaction was isolated as a yellow powder soluble in methanol. Its HR-ESI-MS shows the pseudomolecular ion peak [M + H]+ at m/z 330.0609 a.m.u corresponding to the molecular formula C16H12NO7 (calculated mass for C16H12NO7: 330.0608 a.m.u) contain twelve degrees of unsaturation. The mass difference observed between 1b and the genin of 1 (i.e. 284) is 45 a.m.u, which corresponds to the mass of a nitro group (46 a.m.u) minus one unit (corresponding to the substituted proton). This observation allows us to affirm that 1b comes from the mono-nitration of 1 after hydrolysis. The 1H NMR data of this derivative is close to the substrate 1 but however with some dissimilarities (Table 1). The main difference is the disappearance of the signals attributable to the osidic protons in 1b. We also observe the disappearance of the aromatic H-6 proton signal, resulting in the appearance of the H-8 proton signal as a singlet at δH 6.36 (1H, s).
Similarly, a comparison of its 13C NMR data with that of 1 (Table 2) further confirms the disappearance of the osidic part and the nitration of position 6. Indeed, a concomitant interpretation of the 13C NMR and HSQC spectra of 1b allows us to see that the single aromatic proton of ring A is carried by the carbon at δC 96.0 ppm characteristic of the carbon in position 8 of 5,7-dihydroxylated flavonoids [13] [14] . Indeed, it has been established that in these systems (5,7-dihydroxylated), the proton H-8 is more deshielded than H-6 while its carbon resonates at slightly stronger fields (around 98.0 ppm) than that of H-6 (generally around 100.0 ppm). In addition, the presence of the meta nitro group results in a sight shielding of the C-8 carbon which appears at slightly stronger fields (96.0 ppm). From the aforementioned data, the structure of compound 1b was established as 6-nitrobiochanin A.
Compound 1a obtained from the nitration reaction was isolated as a yellow powder soluble in methanol. Its HR-ESI-MS shows the pseudomolecular ion peak [M + H]+ at m/z 330.0566 a.m.u corresponding to the molecular formula C16H12NO7 (calculated mass for C16H12NO7: 330.0608 a.m.u) contain twelve degrees of unsaturation. Its 1H and 13C NMR data are identical to those of 8-nitrobiochanin A which was previously reported by Al-Maharik and Jaradat in 2020 [12] .
Scheme 2 below shows the plausible mechanism by which derivatives 1a and 1b would have formed [15] .
Step 1. Hydrolysis
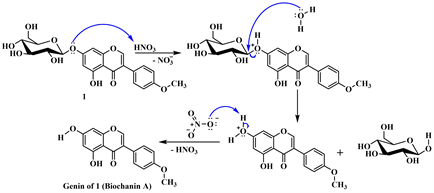
Step 2. Formation of nitronium ion
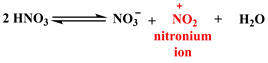
Step 3. Electrophilic attack of nitronium ion and regeneration of nitric acid
![]()
Scheme 2. Mechanistic steps leading to derivatives 1a and 1b.
Compound 1c obtained from acetylation reaction was isolated as yellowish powder soluble in dimethyl sulfoxide. The comparison of its 1H and 13C NMR data with those of substrate 1 (Table 1 and Table 2) shows that the main difference was observed in the sugar moiety. Indeed, in addition to the signal of sugar, we pick up the signals attributed to the proton and carbone of acetyl groups. Compound 1c has been identified at 2",3",4",6"-tetraacetylsissotrin previously synthesized by Ying-Jie and Yi-Cheng in 2009 through reaction between 5,7-dihydroxy-4'-methoxyisoflavone and acetobromoglucose [α-D-glucopyranosyl bromide tetraacetate] [16] .
Scheme 3 below shows the mechanism of the acetylation reaction of 1.
Step 1. Formation of acetylpyridinium acetate
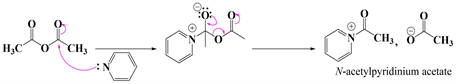
Step 2. Nucleophilic attack
![]()
Scheme 3. Mechanistic steps leading to derivative 1c.
3.2. Antibacterial Activity
The semisynthetic derivatives 1a, 1b and 1c as well as substrate 1 were evaluated for their antibacterial activities (Table 3). This evaluation was carried out on six bacterial strains, three of which were Gram-positive (Staphylococcus aureus ATCC 43300, Methicillin-resistant Staphylococcus aureus 33591 and Staphylococcus aureus CPC) and three Gram-negative (Escherichia coli ATCC 25922, Shighella flexineri CPC and Shighella flexineri NR 518).
Antibacterial cutoff points of pure compounds have been defined as follows: highly active: MIC below 1 µg∙mL−1 (or 2.5 μM), significantly active: 1 ≤ MIC ≤ 10 µg∙mL−1 (or 2.5 ≤ MIC < 25 μM), moderately active: 10 < MIC ≤ 100 µg∙mL−1 (or 25 < MIC ≤ 250 μM), weakly active: 100 < MIC ≤ 1000 µg∙mL−1 (or 250 < MIC ≤ 2500 μM and not active: MIC > 1000 µg∙mL−1 (or >2500 μM) [17] .
Globally, the MIC values ranging from 62.5 µg∙mL−1 to more than 500 µg∙mL−1 depending on compounds and microorganisms. Concerning the substrate 1, from all the strains tested, it shows weak antibacterial activity towards Shighella flexineri CPC (MIC: 250 µg∙mL−1) and Shighella flexineri NR 918 (MIC: 500 µg∙mL−1). It can be seen that derivative 1a was active on all bacteria tested and the best activity (moderate) was observed on Staphylococcus aureus CPC (MIC: 62.5 µg∙mL−1). When tested against on Staphylococcus aureus ATCC 43300 (MIC: 500 µg∙mL−1); Methicillin-resistant Staphylococcus aureus 33591 (MIC: 500 µg∙mL−1); Escherichia coli ATCC 25922 (MIC: 500 µg∙mL−1); Shighella flexineri CPC (MIC: 125 µg∙mL−1) and Shighella flexineri NR 518 (MIC: 250 µg∙mL−1) compound 1a exhibited a weak antibacterial activity and still remain the most active on these strains. All these results are in agreement with the literature which shows that some structural features such as the presence of the sugar moiety or the numbers of hydroxyl, methoxyl groups as well as nitro group had a great influence on their biophysical and biological properties [18] [19] . Unfortunately derivatives 1b and 1c were not active (MIC > 500 µg∙mL−1) against all the strains tested. These observations allow us to state that the hydrolysis followed by the nitration of biochanin A tend to increase the antibacterial
![]()
Table 3. Antibacterial activity (MIC in µg∙mL−1) of 1, 1a, 1b and 1c as well as ciprofloxacin.
/: not determined; S.a: Staphylococcus aureus ATCC 43300; MRSA3391: Methicillin-resistant Staphylococcus aureus 33591; E.c ATCC 25922: Escherichia coli ATCC 25922; S.f CPC: Shighella flexineri CPC; S.f NR 518: Shighella flexineri NR 518; S.a CPC: Staphylococcus aureus CPC; MIC: Minimum Inhibitory Concentration; Ciprofloxacin: drug reference.
position 8 of the isoflavone. They also allow us to suggest that the hydroxyl activity. This increase is also depending on the position of the nitro group (-NO2) because as it seems more pronounced when said nitration takes place at groups found on the osidic part would play a role in the antibacterial activity because their removal (by acetylation) led to a total loss of antibacterial activity (MIC ˃ 500 µg∙mL−1) compared to the substrate (MIC: =250 and 500 µg∙mL−1).
4. Conclusion
The present work describes the semisynthesis and structural elucidation of three derivatives (1a, 1b and 1c) obtained from an isoflavone, sissotrin (1) isolated from Trifolium baccarinii after nitration and acetylation reactions. The results of the evaluation of their antibacterial properties showed that only derivative 1a exhibited moderate activity (62.5 µg∙mL−1) on Staphylococcus aureus CPC. The substrate 1 showed only weak activity against Shighella flexineri CPC and Shighella flexineri NR 518. Based on these results, we can recommend that more such transformations on isoflavones be carried out in order to better understand and establish the structure-activity relationships.
Acknowledgements
The authors gratefully acknowledge Billy Toussie Tchegnitegni (University of Dschang), Pierre Mkounga (University of Yaounde I) and Augustin Ephrem Nkengfack (University of Yaounde I) to provide the facilities needed to carry out this work. The “Institut de Chimie Moléculaire de Reims” and the University of Dschang are gratefully acknowledged for administrative and technical support.