Correlation between Pubertal Delay in Adolescents with Homozygous Sickle Cell Disease and Socio-Demographic, Clinical Factors ()
1. Introduction
Pubertal development represents a physical, biological and psychological process, which results in the acquisition of reproductive capacities [1] . Abnormalities of pubertal development constitute a frequent reason for consultation [2] . Pubertal development is regulated by activating and inhibitory factors. Among the factors inhibiting pubertal development are chronic diseases including sickle cell anemia, the leading hemoglobinopathy in the world and which is a public health problem [3] .
Several studies report that sickle cell disease is responsible for endocrine complications in adolescence, including growth retardation and pubertal delay [4] [5] .
Hospital statistics for pubertal delay in adolescents living with homozygous sickle cell disease vary worldwide, ranging from 8% to 50% in Europe and 15% in Latin America [5] [6] . In Africa, the reported data confirm the same trends, particularly in Egypt: 25% and Nigeria: 75% [7] [8] .
In Congo, studies carried out on patients living with homozygous sickle cell disease show high prevalence of delayed puberty ranging from 28.7% to 53.7% and therefore constitutes a worrying health problem [9] [10] .
Some factors influencing pubertal development in adolescents living with homozygous sickle cell disease such as inadequate nutritional intake and deficiencies in essential micronutrients are preventable [11] [12] . Several questions remain open, namely how many Congolese adolescents living with homozygous sickle cell disease are vulnerable to good pubertal development and what are the conditions, or even the internal and/or external predispositions (factors) which are unfavorable or protective for normal pubertal development.
It is therefore necessary for these factors to be known to practitioners in order to integrate them into the overall management of sickle cell disease. It is in this perspective that we set ourselves the general objective of studying the pubertal development of adolescents living with homozygous sickle cell disease in Brazzaville, and more specifically:
1) Describe the sociodemographic and clinical characteristics of adolescents living with homozygous sickle cell disease in Brazzaville;
2) Report the prevalence of abnormalities of pubertal development in adolescents living with homozygous sickle cell disease in Brazzaville;
3) Identify the sociodemographic and clinical factors that delay the pubertal development of adolescents living with homozygous sickle cell disease in Brazzaville.
2. Patients and Methods
This was a multicenter analytical cross-sectional study with prospective data collection. The study took place from April 1st to October 30th, 2022, in seven months. The study was conducted in the following health centers: The National Reference Center for Sickle Cell Disease, the Pediatrics Department of the Brazzaville Hospital and University. During the study period, 982 patients living with sickle cell disease were seen on an outpatient or inpatient basis at the selected health facilities, including 353 adolescents. The inclusion criteria were: only adolescents, in accordance with the WHO definition of an adolescent as any person aged between 10 and 19 years [13] , The inclusion criteria were: only adolescents according to the WHO definition which considers as adolescents any person between the ages of 10 and 19 years [13] , having performed a blood count and received prior consent from the parent/guardian for minors or having consented to participate in the study for adults. Exclusion criteria among adolescents were: another known chronic pathology (congenital heart disease, respiratory insufficiency, HIV/AIDS infection, endocrinopathies) that may affect growth; living adolescents with homozygous sickle cell disease in a gestational state. 6 adolescents were excluded for a total of 347 adolescents participating in the study.
The data was collected using a survey form (Appendix I) including a questionnaire on the sociodemographic characteristics of the adolescent, the natural history of sickle cell disease (completed by the adolescent’s follow-up form or diary) and the age of onset of certain secondary sexual characteristics; A physical examination including Anthropometric measurements including weight and height for assessment of nutritional status and growth. Nutritional status was assessed by calculating body mass index (BMI) and height/age and weight/age ratios. Wasting: when the BMI/A ratio, relative to the WHO standard deviation curves, is between −3DS and −2DS, Severe wasting: when the BMI/A ratio, relative to the WHO standard deviation curves, is below −3DS.
All these measurements were related to the new WHO growth curves (Appendix II). Assessment of pubertal development using the Tanner classification (Appendix III). This study was carried out after obtaining informed consent from the parents and the agreement of an ethics committee (Appendix IV) in accordance with the Helsinki declaration. The variables related to the adolescent were sociodemographic: age (in year and month), sex, social status (orphan or not), diet (number of meals per day); and clinical: circumstances of discovery of the disease, number of hospitalizations/year, number of transfusion episodes since discovery of the disease, status of acute and chronic complications, splenectomy, nutritional status: (P, T, BMI); age of onset of secondary sexual characteristics (thelarche, menarche, pubarche, spermache, gynecomastia); pubertal stage according to Tanner’s Classification. The survey data were entered using Microsoft Excel version 2013. SPSS version 20 was used to process and analyze the data. Central tendency parameters were used to describe the quantitative variables. For the univariate analysis, the significance threshold of 5% was considered in the search for potential risk factors. Thus a correlation exists between two variables when the p-value is less than or equal to 0.05.
3. Results
In our study, 347 adolescents living with homozygous sickle cell disease were registered out of 353 homozygous sickle cell adolescents during the study period. The participation rate was 98.3%. The adolescents were on average aged 15.1 ± 2.5 years with extremes of 10 and 19 years. Their distribution according to age groups (in years) is recorded in Table 1.
At the end of the study, 182 (52.4%) adolescents were male and 165 (47.6%) female, i.e. a sex ratio = 1.1
Fifty-six (16.1%) adolescents selected for the study were orphans, 48 of whom were single-parent orphans and 8 were two-parent orphans. The diet of adolescents living with sickle cell disease was varied for 346 (99.7%) of them. One adolescent (0.3%) was vegetarian. The average number of meals per day was 2.59 ± 0.5 with extremes of 1 and 4 meals per day.
One hundred and forty adolescents (40.6%) consumed less than 3 meals per day.
All adolescents included in this study had already been hospitalized at least once, 19.3% of whom had more than five hospitalizations per year.
The average number of hospitalizations per year was 4.15 ± 1.8 with extremes of 1 and 12 hospitalizations/year. Three hundred and twenty-six adolescents (93.9%) had already been transfused. The average number of blood transfusions was 3.7 ± 2.6 with extremes of 1 and 18 transfusions.
The mean age at the time of the first blood transfusion was 5.07 ± 3.2 years with extremes of 18 months and 17 years. In our study the main causes of
![]()
Table 1. Distribution of adolescents by age groups.
n: partial workforce; %: percentage.
hospitalizations were vaso-occlusive and anemic crises. The distribution of adolescents according to the type of acute complications is shown in Table 2. Adolescents who had vaso-occlusive crises numbered 345. The number of episodes of these bone CVOs was less than or equal to 3 in 240 cases (69.6%); in 105 cases (29.8%), it was greater than 3.
Adolescents presented with acute chest syndrome in 29 cases (8.4%). Priapism was observed in 54 cases (29.7%). The strokes were all ischemic, and observed in 18 cases (5.2%). The distribution of adolescents according to the type of chronic complications is shown in Table 3. It is mainly gallbladder lithiasis in 145 cases (41.8%), followed by osteonecrosis of the femoral head in 43 cases (12.4%).
One hundred and fifty-two (43.8%) adolescents had good nutritional status (Figure 1). Stature growth was normal in 192 cases (55.3%) and 155 (44.7%) adolescents had growth delay.
Growth retardation was severe in 55 (15.9%) adolescents with a T/A ratio lower than minus 3 SD according to new WHO growth standards.
In girls, the average age of onset of breast development, also called thelarche, was 13.09 ± 1.7 years with extremes of 10 and 16 years. One hundred and eleven
![]()
Table 2. Distribution of adolescents by type of acute complications.
n: Partial workforce; %: Percentage; CVO: Vaso-occlusive crisis; ATS: Acute chest syndrome; Stroke: Acute vascular accident.
![]()
Table 3. Distribution of adolescents by type of chronic complications.
n: partial workforce; %: percentage.
adolescents (67.3%) had already started puberty.
The average age of onset of menarche was 15.95 ± 1.8 years with a range of 13 and 18 years. During the study period, fifty-six adolescent girls (33.9%) already had their first menstruation.
In boys, the average age of appearance of spermarche was 15.76 ± 1.3 years with extremes of 13 and 18 years. Forty-six adolescents (13.3%) had already had their first ejaculation.
In both sexes, the average age of pubarche was 13.91 ± 1.7 years with extremes of 10 and 18 years. During our study, 196 adolescents (56.5%) had normal pubertal development for their age. An abnormality of pubertal development was found in 151 adolescents, or 43.5%.
The distribution of adolescents according to pubertal development is shown in Figure 2.
From sociodemographic point, among the 54 orphan adolescents, those with delayed puberty (34) were significantly more numerous (Table 3) than those
![]()
Figure 1. Distribution according to nutritional status of adolescents with homozygous sickle cell disease.
![]()
Figure 2. Distribution according to pubertal development of adolescents with homozygous sickle cell disease.
with normal puberty, numbering 20 (p = 0.00127). Under-nutrition in our series was correlated with pubertal delay (Table 3), since among the 137 adolescents having less than 3 meals/day, 80 had pubertal delay compared to 57 with normal puberty (p = 0.0000).
Clinically in our series, frequent hospitalizations (more than 5 hospitalizations/year) contributed to pubertal delay (Table 4), since pubertal delay was significantly more observed in 39 cases compared to 25 cases with normal puberty among hospitalized adolescents more than 5 times/year (p = 0.0013). pubertal delay was significantly greater (p = 0.0000) than cases of normal puberty in the category of adolescents having had more than 3 vaso-occlusive crises/year. Blood transfusion allowed normal puberty in 176 adolescents compared to 147 with delayed puberty (p = 0.0002).
However, when the transfusions were frequent (more than 5 since the discovery of the disease), pubertal delay was significantly more reported in 37 cases, compared to 26 with normal puberty (p = 0.0127) (Table 4).
Among the adolescents who experienced priapism (Table 5), 28 had delayed puberty compared to 24 with normal puberty (p = 0.04). Wasting also contributed
![]()
Table 4. Correlation between pubertal delay and clinical aspects.
PD: Pubertal Delay; NP: Normal Puberty; OR: Odds ratio; CI: Confidence interval; * Reference CVO: Vaso-occlusive crisis.
![]()
Table 5. Correlation between pubertal delay and clinical aspects (2).
ATS: Acute chest syndrome; ONTF: Osteonecrosis of the femoral head, OMC: Chronic osteomyelitis.; SMG: splenomegaly.
to pubertal delay, since in the group of wasted adolescents, pubertal delay was observed in 113 cases compared to 80 cases of normal puberty (p = 0.0000). (Table 5)
4. Discussion
To carry out this study, a total of 347 adolescents were selected. Our sample size is larger than that of two previous studies carried out in Brazzaville by Mpemba Loufoua et al.: 72 cases and 53 cases respectively [9] [14]
The average age of the adolescents was 15.1 ± 2.5 years. Adolescents aged 16 to 19 are the most represented; our results are similar to those of Adeyemo et al. in Nigeria who reported a mean age of 16.0 ± 1.5 years in their series [15] . The predominance of boys reported by Minto’o Rogombé et al. in Gabon and Bah et al. in Mali was confirmed by this study, in which boys were the most represented [16] [17] . However, Simo et al. in Congo and Aloni et al. in the DRC reported a predominance of females [18] [19] . On this, sickle cell anemia is a genetic disease without gender predominance due to its autosomal transmission and its recessive nature. The differences observed between surveys could be linked to the demographics of each survey location or even to recruitment biases.
Orphaned adolescents represented 16.1% of the study population, contrary to the data reported by Ollandzobo et al.: 32.5%. [20] . The methodological difference is surely the most plausible explanation.
The majority of adolescents in this study (51%) were followed by the national sickle cell reference center (CNRDr). These results illustrate the interest of this specialized center in our health system, more specifically in the management of sickle cell disease. Vaso-occlusive and anemic crises are the main causes of hospitalization of adolescents living with homozygous sickle cell disease in Brazzaville in 99.4% and 93.9% respectively. This trend has already been reported in Brazzaville by Mbika Cardorelle et al.: 52.5% and 47% respectively [21] . Chronic complications were dominated by gallstone lithiasis (41.8%). This result is similar to that reported by Ngolet et al. where gallbladder lithiasis represented 40.3%. [22]
Adolescents with sickle cell disease have normal pubertal development (56.5%). Mandese et al. in Italy, report a prevalence of 50% [5] . The improving therapeutic progress in management of sickle cell anemia as well as the establishment of specialized centers in recent decades have improved the pubertal development of adolescents.
Furthermore, the prevalence of abnormalities of pubertal development was 43.5%, including 42.6% pubertal delay and 0.9% impuberty. The prevalence of pubertal delay in our study is lower than that reported by Mabiala Babela et al.
in Congo which reports a prevalence of 53.7% [10] . The improvement in monitoring and ECP through the intensification of treatment with the use of hydroxyurea in recent years in Congo may explain the improvement in pubertal development in adolescents living with sickle cell disease. However, efforts are still needed to reduce the prevalence of this pubertal delay to lower levels as reported by Özen in Turkey: 8% [4] .
In our series, delayed puberty affected 21.5% of boys and 21.1% of girls. In Congo, the studies carried out report prevalence higher than those of ours: 30.2% and 42% respectively among boys and girls [9] [14] . This disparity could be justified by improvement in the care of patients living with sickle cell disease over the years. In fact, Mpemba-Loufoua’s studies date back more than two decades as well as the small size of the sample.
As for impuberty, it was found in 0.9% of our study population, only among boys. Some authors consulted reported prevalence higher than ours varying from 25% to 75% [7] [8] [14] . The size of the sample as well as the methodology of each study would justify this disproportion. Nevertheless, there is consistency in the results on the predominance of gender; men are more immature than girls.
Puberty began on average at the age of 13.09 years in girls and menarche appeared on average at the age of 15.9 years. Our results are consistent with those described in the literature: the average age of onset of puberty is delayed by two years compared to adolescents without sickle cell disease [23] [24] [25] . On the other hand, the data reported in Italy indicate an average age of menarche of 12.8 years [5] . This discordance could be justified by the secular advance but also the genetic, nutritional and psychosocial factors of the populations. The pubertal delay estimated at 42.6% in this work, is associated with several factors including: under-nutrition with a number of daily meals less than three, the number of hospitalizations greater than 5 per year.
Under-nutrition is a factor hindering the growth and development of Congolese adolescents: 40.6% in this study. According to the Congo Demographic Health Survey (EDS) 2012, 23% of households have inadequate food consumption, preventing them from leading a healthy and active life [26] .
Similarly, in its 2018 annual report, the United Nations Children’s Fund (UNICEF), reports that 149 million children worldwide are stunted, nearly 50 million are wasted and more than 340 Million suffer from a deficiency in essential micronutrients, also known as “hidden hunger” [27] . Thus, under-nutrition associated with sickle cell anemia, a hypermetabolic pathology due to the increased energy expenditure it requires, hinders the maturation of organs and therefore pubertal development.
The number of hospitalizations beyond five constitutes in our study a factor associated with RP. Nineteen percent of adolescents had more than 5 hospitalizations/year. M’pemba-Loufoua in his series reported the average number of hospitalizations greater than 5 per year was statistically significant (p = 0.033) [14] . Frequent hospitalization is often a reflection of the severe form of the disease; it is caused by episodes of recurrent CVO and frequent deglobulization crises leading to anorexia and psychological distress with the consequence of having an impact on growth and pubertal development. Our study had experienced pitfalls including the lack of funding which did not allow the realization of biological explorations such as hormonal balances that could have informed the dynamics of these parameters during puberty in adolescents living with homozygous sickle cell disease. The diagnosis of puberty was therefore only clinical, based on Tanner’s classification.
Also, the transversal nature of the study gives only a reflection of an instantaneous photograph. A long-term longitudinal study would be wise and preferable to determine the dynamics of puberty in sickle cell adolescents as it is a process.
5. Conclusions
The study of the pubertal development of Congolese adolescents living with homozygous sickle cell disease confirmed the hospital statistical variabilities observed throughout the world. These variations could be explained by the fact that the factors hindering pubertal development are intrinsic (sickle cell disease with its clinical forms, its complications) and extrinsic (environmental: diet, social status).
The onset of secondary sexual characteristics is delayed by an average of two years compared to the general population. The factors associated with pubertal delay in our context are: fewer than three meals a day, a number of hospitalizations greater than 5 per year, Systematic screening of these associated factors during the follow-up of prepubertal children living with homozygous sickle cell disease is required in order to establish a prognostic score for delayed puberty.
Appendix I: Survey Form
SURVEY SHEET
SHEET N˚/____/ DATE OF SURVEY___/___/___
I. Identify
Gender: Male /__/ Female /__/
Date of Birth: ___/__/_____ Age: /____/ years
Level of education: Primary /__/ Secondary 1st degree /__/ Secondary 2nd degree /__/ Higher /__/ Not at school /__/
Residence: Makélékélé /__/ Bacongo /__/ Poto-Poto /__/ Moungali /__/ Ouenzé /__/ Talangaï /__/
Mfilou /__/ Madibou /__/ Djiri /__/ Mbamou Island /__/
Carergiver: Father /__/ Mother /__/ Guardian /__/ (Specify_______________)
Social Status: Orphan /__/ Not orphan /__/ If yes: Father /__/ Mother /__/ Two-parent /___/
Consultation center: CNRD /__/ PGE /__/ CME /__/
II. History
1. Natural history of sickle cell disease
- Age at diagnosis: 1st year of life /__/ 1 - 5 years /__/ 6 - 10 years /__/ ≥10 years/__/
- Circumstance of discovery: Screening /__/ CVO /__/ Anemic crisis /__/ Hand-foot syndrome/__/ Other (Specify_________________________________)
- Electrophoretic profile: % HbS /___/ % HbA2 /___/ % HbF /___/ % Other /______/
- Baseline hemoglobin level: /_________/ g/dl
- Acute complications:
o Number of CVOs per year: /___/
o Number of anemic attacks since birth: /___/
o Blood transfusion: Yes /__/ No /__/
If yes, Age of first TS /_____/(months/years) Number since birth: /___/
o Has he ever had a stroke: Yes /__/ No /__/
If yes, Number since discovery of the disease: /__/ Type: /______________/
o Has he ever had acute chest syndrome: Yes /__/ No /__/
If yes, number since discovery of the disease: /__/
o Has he ever had priapism: Yes /__/ No /__/
If yes, number since discovery of the disease: /__/
- Chronic complications:
o Heart failure: Yes /__/ No /__/
o Osteonecrosis of the femoral head: Yes /__/ No /__/
o Chronic osteomyelitis: Yes /__/ No /__/
o Leg ulcer: Yes /__/ No /__ /
o Gallbladder lithiasis: Yes /__/ No /__ /
o Nephropathy: Yes /__/ No /__ /
- Splenectomy: Yes /__/ No /__/
- Hospitalization: Yes /__/ No /__/
If Yes, average number of hospitalizations per year: /__/
- Monitoring quality: Regular /__/ Irregular /__/
- Maintenance treatment:
o Folic acid: Yes /__/ No /__/
If yes, Regular /__/ Irregular /__/
Number of tablets/day /___/
o Hydroxy urea: Yes /__/ No /__/
If yes,
Indication:/______________________________/ Start date:/______________/
o Transfusion Exchange: Yes/__/ No /___/
If yes, Indication: /________________________/ Number: /___/
- Marrow transplant: Yes/___/ No /___/
If yes, When: /___/
- Vaccination:
o Thyphim VI Yes /__/ No /__/
o Pneumo23 Yes /__/ No /__/
o Meningococcus Yes /__/ No /__/
o Anti-influenza Yes /__/ No /__ /
2. Other personal history
- Thelarche (in girls) Yes/__ / No/__/
o If yes, age of onset: /___/ years
- Menarche (in girls): Yes /__/ No /__/
o If yes, age of onset: /___/ years
- Spermarch (in boys): Yes /__/ No /__/
o If yes, age of onset: /___/ years
- Pubarche: Yes /__/ No /__/
o If yes, age of onset /___/ years
- Food :
o Diet: Normal (varied) /__/ Vegetarianism /__/ Veganism /__/
o Number of meals/day: 1 /__/ 2 /__/ 3 /__/
- Drinking alcohol: Yes /__/ No /__/
3. Family history
- Size of Sibling: /__/ Position in Sibling: /__/
- Number of other homozygous sickle cell children: /__/
- Marital status of parents: Couple /__/ Single /__/ Recomposed /__/
Socio-economic level: Low /__/ Medium /__/ High /__/
III. Physical Examination
1. Anthropometric measurements
- Weight: /___/ Kg Height: /___/ m BMI: /____/ Kg/m 2
- T/A: /____/ SD Either
- Normal /___/
- Growth retardation /___/
- Significant growth retardation /___/
- BMI / A: /___/ SD Either
- Normal /___/
- Wasting /___/
- Severe wasting /___/
- Overweight /___/
- Obesity /___/
2. Coloring of the mucous membranes
- Normal /__/ Pallor: Slight /__/ Moderate /__/ Clear /__/
3. Jaundice: Yes /__/ No /__/ If yes, Mild /__/ Moderate /__/ Frank /__/
4. Splenomegaly: Yes /__/ No /__/ If yes, Hackett stage: I; II; III; IV
Appendix II: Growth Indicators According to WHO (Z Score)
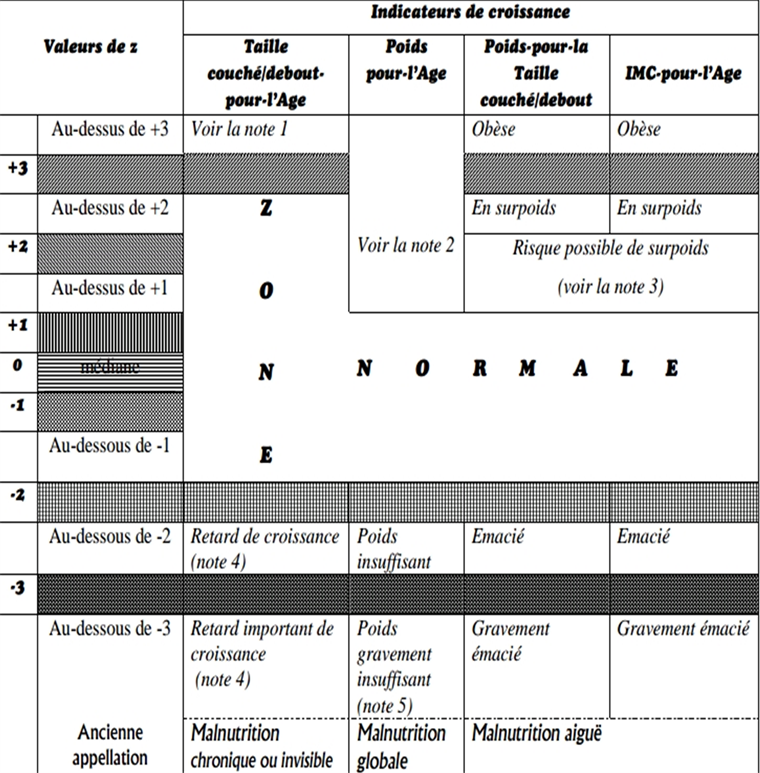
Appendix III: TANNER classification
Girl: S /__/ S /__/ Boy: P /__/ G /__/
Precocious puberty /__/ Pubertal delay /__/ Normal puberty /__/ Impuberty /__/
For the girl:
S1P1: Impubescent stage
S2P2: The average age is around 10½ to 11 ± 2 years
S3P3: The average age is around 12 ± 2 years
S4P4: The average age is around 13 ± 2 years
S5P5: The average age is around 14½ ± 3 years
In the boy:
G1P1: Impubescent stage
G2: The average age is around 11½ ± 1 years & P2, the average age is 13½ years
G3: The average age is around 12½ ± 2 years & P3, the average age is 14 years
G4: The average age is around 13½ ± 2 years & P4 the average age is 14½ years
G5P5: The average age is around 15 ± 2 years