Investigation into Protective Groups for Anilino Functionality upon Oxone-Mediated Oxidative Esterification ()
1. Introduction
In the course of organic synthesis, particularly for multi-step synthesis or natural product total synthesis, selecting the appropriate protective groups for the intended functionalities is crucial in order to achieve chemoselective synthetic goals. For syntheses that often require protection, the development of many protective groups has been based on compatibility with the functionality of the anilino group. Indeed, there are a number of protective units available for anilino groups, and many of the preparation methods have been introduced over the past decades [1] . Among them, the most common and versatile protective groups for anilino functionality include alkyl groups, amides, carbamates, and sulfonamides.
Today, there are a few methods for oxidative esterification that transform aldehydes to their corresponding carboxylic esters. Reactions using tert-butyl hydroperoxide (TBHP) as an oxidant and tris(pentafluoropheny)borane as a catalyst have been reported, and para-aminobenzaldehyde could provide its methyl ester without any observable side reactions with the amino group [2] . Also, oxidative esterification over anchored phosphotungstates has been introduced as a method to achieve conversion from para-aminobenzaldehyde to a methyl ester [3] . Visible light-assisted esterification employing a cobalt complex grafted to nanoporous graphitic carbon nitride is used to transform meta-aminoben-zaldehyde into a methyl ester [4] . Other examples of ester production that begin with para-aminobenzaldehyde derivatives utilize TBHP with Bu4NI as a catalyst [5] or TBHP with Cu(OAc)2 as a catalyst [6] . Photoinduced acylation in the presence of iridium and nickel bromide catalysts has been developed to furnish a variety of esters [7] , and dehydrogenative coupling by a nickel hydride complex is used to simultaneously produce esters and alcohols [8] . However, in all these cases, the protective groups for anilino functionality are reportedly limited only to acetyl (Ac) and dialkyl groups.
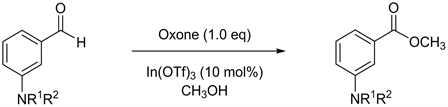
Meanwhile, we previously conducted research on indium metal and developed a mild methodology wherein deprotection of the trichloroethoxycarbonyl (Troc) moiety released free amine and aniline products [9] , as well as alcohol and phenol products [10] . Also, we recently reported practical methodologies for the oxidative esterification of aromatic aldehydes using Oxone® monopersulfate compound (Oxone) as an oxidant and indium(III) triflate as a catalyst [11] [12] [13] [14] . Based on our continuous study, compatibility of protective groups with anilino functionality was investigated via the implementation of Oxone-mediated oxidative esterification in methanol. To the best of our knowledge, no comprehensive study has dealt with oxidative esterification of aldehydes that possess anilino functionalities. The results and the details of our experiments are reported herein.
2. Results and Discussion
2.1. Unstable Groups under the Reaction Conditions
In our previous study, Oxone-mediated oxidative esterification in methanol was examined using meta-hydroxybenzaldehyde derivatives as the starting materials. Through these practical experiments, the stable protective groups and unstable protective groups for phenolic functionality under the reaction conditions were identified [14] . In a continuation of our study, we then subjected meta-amino-benzaldehyde derivatives to Oxone-mediated oxidative esterification to determine their compatibility with anilino functionality.
First, we envisioned derivatives protected by tert-butoxycarbonyl (Boc) [15] and benzyl (Bn) [16] groups. These are the popular examples, because they are known as convenient and versatile protective groups. Both starting materials were then subjected to Oxone-mediated oxidative esterification in methanol, but neither furnished the expected products. Removal of the Boc protection from the starting material resulted in a 13% yield of methyl ester with a free amino group (Table 1, Entry 1). The Bn-protected starting material showed no conversion into its methyl ester. Instead, this starting material was merely recovered in a 23% yield (Table 1, Entry 2). We then extended our interest to the methyl protection, since the methyl group is known as a simple alkyl protective unit for the anilino moiety [17] [18] . However, despite our expectations, neither monomethyl nor dimethyl protection could not stabilize the Oxone-mediated
![]()
Table 1. Esterification reactions using the starting materials with unstable groups.
aIsolated yields.
esterification reactions, which ended in decompositions (dec) (Table 1, Entries 3 and 4). In another attempt we employed diallyl protection [19] , but there was no reaction (NR) without the corresponding methyl ester (Table 1, Entry 5).
2.2. Stable Groups under the Reaction Conditions
The Boc group is often removed due to its sensitivity to acid. Therefore, acid-resistant carbamates such as Troc [20] [21] and allyloxycarbonyl (Alloc)-protected starting materials [22] were prepared and subjected to Oxone-mediated oxidative esterification. In both cases, the reactions proceeded smoothly. According to our previous study, the electron withdrawing groups should display more positive effects than the electron donating groups [13] . We suggest that the strong electron withdrawing properties of the Troc group resulted in more of an excellent quantitative yield compared with that of the Alloc groups (Table 2, Entries 1 and 2). Amides also possess electron withdrawing characteristics. Consequently, we performed reactions with starting materials that possess typical amides such as benzyl (Bz) and Ac [23] . Again, in both cases, the reactions proceeded smoothly and gave both a quantitative yield and a 69% yield, respectively (Table 2, Entries 3 and 4) [24] . The aromatic Bz group, due to its aromaticity, seemed to stabilize the reaction process very well. Substituting a trichloroacetyl group [23] [25] for an Ac group slightly improved the yield (Table 2, Entry 5). Finally, we also explored a couple of sulfonamide derivatives [26] , which are known to possess powerful electron withdrawing effects. The nosyl (Ns) group has an electron withdrawing nitro structure at the 2-positon on the benzene ring, and the presence

![]()
Table 2. Esterification reactions using the starting materials with stable groups.
aIsolated yields.
of two Ns groups was found to be more productive than one Ns group (Table 2, Entries 6 and 7). Furthermore, the phenylsulfonyl group has no substituents, whereas the tosyl (Ts) group has an electron donating methyl structure at the 4-positon on the benzene ring. When Ns (Table 2, Entry 7), phenylsulfonyl (Table 2, Entry 8), and Ts structures (Table 2, Entry 9) are compared, their yields could be explained as the result of electronic influence when they all are doubly substituted. The yield was the highest with the two Ns groups, and it was lowest with the two Ts groups.
3. Conclusion
Protective groups were investigated for compatibility with anilino functionality by utilizing the results of our previous study. Oxone-mediated oxidative esterification in methanol was carried out to determine the compatibility of groups used to protect anilino functionality. Under the reaction conditions, alkyl protective groups were totally unstable and their use resulted in no products. The Boc group did not hold under the reaction conditions and thus was cleaved from the anilino moiety. On the other hand, starting compounds with other carbamates, as well as amides and sulfonamides, as the protective groups for the anilino functionality, successfully furnished the corresponding methyl esters in good to excellent yields with the protective moieties intact. The results of this study indicate that the electron withdrawing properties of the protective parts have a positive effect on reaction productivity.
4. Experimental
4.1. Materials and Instruments
All reagents were of analytical grade, were purchased commercially, and were used without further purification. All reactions were performed under argon using magnetic stirring unless otherwise stated. 1H NMR and 13C NMR spectral data were recorded on a JEOL JMTC-500 spectrometer (500 MHz for 1H NMR and 125 MHz for 13C NMR) using tetramethylsilane (TMS) as the internal standard.
4.2. General Experimental Procedure
When used as the starting materials, benzaldehyde derivatives such as meta-tert-butoxycarbonylamino benzaldehyde (111 mg, 0.5 mmol) were combined with Oxone (308 mg, 0.5 mmol) and indium (III) triflate (28 mg, 10 mol%) in methanol (25 mL). The reaction mixtures were heated to 50˚C and monitored for completion via TLC. The reaction mixtures were filtered, and the filtrate was condensed via rotary evaporation. The resultant residue was purified by silica gel flash column chromatography to obtain the methyl ester products, which were concentrated via rotary evaporation and dried using a vacuum pump overnight. The yields reported are the isolated yields. All products were confirmed by spectroscopy.
Methyl 3-(2,2,2-Trichloroethoxycarbonylamino)benzoate (Table 2, Entry 1): 1H NMR (500 MHz, Chloroform-d) δ 8.05 (t, 1H, J = 1.8 Hz), 7.80 - 7.77 (m, 2H), 7.43 (t, 1H, J = 7.5 Hz), 7.17 (br s, 1H), 4.84 (s, 2H), 3.93 (s, 3H); 13C NMR (125 MHz, Chloroform-d) δ 166.6, 151.5, 137.4, 131.1, 129.3, 125.2, 123.2, 119.9, 95.1, 74.6, 52.3.
Methyl 3-(Allyloxycarbonylamino)benzoate (Table 2, Entry 2): 1H NMR (500 MHz, Chloroform-d) δ 8.03 (s, 1H), 7.80 (d, 1H, J = 5.7 Hz), 7.72 (d, 1H, J = 8.0 Hz), 7.36 (t, 1H, J = 8.0 Hz), 7.34 (br s, 1H), 5.98 - 5.90 (m, 1H), 5.34 (dd, 1H, J = 17.2, 1.7 Hz), 5.24 (dd, 1H, J = 10.3, 1.2 Hz), 4.66 (d, 2H, J = 5.7 Hz), 3.89 (s, 3H); 13C NMR (125 MHz, Chloroform-d) δ 166.9, 153.3, 138.3, 132.2, 130.7, 129.1, 124.3, 123.0, 119.6, 118.3, 65.9, 52.3.
Methyl 3-(2,2,2-Trichloroacetamido)benzoate (Table 2, Entry 5): 1H NMR (500 MHz, Chloroform-d) δ 8.59 (br s, 1H), 8.14 (t, 1H, J = 1.7 Hz), 7.90 (dd, 1H, J = 8.0, 2.3 Hz), 7.87 (d, 1H, J = 8.0 Hz), 7.46 (t, 1H, J = 8.0 Hz), 3.91 (s, 3H); 13C NMR (125 MHz, Chloroform-d) δ 166.2, 159.5, 136.2, 131.1, 129.4, 127.0, 124.8, 121.5, 92.5, 52.4.
Methyl 3-(ortho-Nitrobenzenesulfonamide)benzoate (Table 2, Entry 6): 1H NMR (500 MHz, Acetone-d6) δ 7.99(dd, 1H, J = 8.0, 1.2 Hz), 7.98 - 7.96 (m, 2H), 7.89 (dt, 1H, J = 8.0, 1.2 Hz), 7.81 - 7.78 (m, 2H), 7.58 (ddd, 1H, J = 8.0, 2.3, 1.2 Hz), 7.47 (t, 1H, J = 8.0 Hz), 3.87 (s, 3H); 13C NMR (125 MHz, Acetone-d6) δ 166.6, 149.4, 138.0, 135.7, 133.4, 132.6, 132.4, 132.1, 130.6, 127.15, 127.08, 126.0, 123.4, 52.6.
Methyl 3-bis(ortho-Nitrobenzenesulfonamide)benzoate (Table 2, Entry 7): 1H NMR (500 MHz, Acetone-d6) δ 8.38 (dd, 2H, J = 8.0, 1.7 Hz), 8.17(dt, 1H, J = 7.5, 1.7 Hz), 8.11 (dt, 2H, J = 8.1, 1.8 Hz), 8.03 (dt, 2H, J = 8.0, 1.2 Hz), 7.95 (dd, 2H, J = 8.0, 1.2 Hz), 7.92 (t, 1H, J = 1.7 Hz), 7.62 (t, 1H, J = 8.0 Hz), 7.59 (dt, 1H, J = 8.0, 1.7 Hz), 3.88 (s, 3H); 13C NMR (125 MHz, Acetone-d6) δ 166.0, 149.4, 137.9, 137.7, 134.1, 133.6, 133.44, 133.37, 132.7, 132.5, 130.8, 130.7, 125.6, 52.8.
Methyl 3-bis(Benzenesulfonamide)benzoate (Table 2, Entry 8): 1H NMR (500 MHz, Chloroform-d) δ 8.13 (dt, 1H, J = 8.1, 1.2 Hz), 7.94 - 7.92 (m, 4H), 7.71 - 7.68 (m, 3H), 7.58 - 7.55 (m, 4H), 7.44 (t, 1H, J = 8.0 Hz), 7.21 (ddd, 1H, J = 8.0, 2.3, 1.2 Hz), 3.89 (s, 3H); 13C NMR (125 MHz, Chloroform-d) δ 165.6, 139.2, 135.8, 134.5, 134.2, 132.6, 131.6, 131.3, 129.3, 129.1, 128.6, 52.4.
Methyl 3-bis(para-Toluenesulfonamide)benzoate (Table 2, Entry 9): 1H NMR (500 MHz, Chloroform-d) δ 8.12 (dt, 1H, J = 8.1, 1.2 Hz), 7.80 (d, 4H, J = 8.6 Hz), 7.73 (t, 1H, J = 1.7 Hz), 7.43 (t, 1H, J = 8.0 Hz), 7.34 (d, 4H, J = 8.1 Hz), 7.20 (ddd, 1H, J = 8.1, 2.3, 1.2 Hz), 3.90 (s, 3H), 2.48 (s, 6H); 13C NMR (125 MHz, Chloroform-d) δ 165.7, 145.2, 136.4, 135.9, 134.8, 132.7, 131.5, 131.2, 129.7, 129.2, 128.6, 52.4, 21.7.