Childhood Myopathy: Service Experience Neurology at Ignace Deen National Hospital and Simbaya Neurological Institute ()

1. Introduction
Neuromuscular diseases are ubiquitous diseases and therefore present in all ethnic communities and at all latitudes [1].
Progressive forms appearing in children include Duchenne muscular dystrophy (DMD) which is an X-linked recessive disease affecting between 1/3600 and 1/6000 male live births. Spinal muscular atrophy (SMA) is an autosomal recessive disease with a spectrum of disease severity, and a number of other neuromuscular conditions [2].
Childhood muscular dystrophy (MDE) is rarely reported in Africa, particularly in its sub-Saharan region [3]. Rare studies published in South Africa (1994) showed a prevalence of 1/100,000 for Duchenne muscular dystrophy and 1/755,000 for Becker muscular dystrophy [4].
However, a study carried out by the French Society of Myology on myopathies in West Africa showed an overall molecular confirmation rate of 45.3% [5].
In Guinea, to our knowledge, no studies on these conditions in children have been found.
The symptomatology of MDE is dominated by the classic myogenic syndrome characterized by: muscle weakness, myalgia and cramps, proximal and bilateral motor deficit, amyotrophy or more rarely hypertrophy, hypotonia, hyporeflexia, tendon retractions [6].
The most widely used diagnostic tools in patients suspected of myopathy are: serum creatine phosphokinase (CPK) assay, electroneuromyogram (ENMG), muscle biopsy, magnetic resonance imaging (MRI) and blood. Molecular genetics has DNA extraction [5].
In the absence of curative treatment, the current management of these chronic pathologies is essentially palliative. However, improving the medical follow-up of these patients can increase their life expectancy. Myopathic patients are highly dependent on those around them. No study has focused on measuring the health-related quality of life of caregivers who regularly care for these people [7].
The objective of this study was to describe the clinical, paraclinical and therapeutic aspects of children with myopathies and to assess the quality of life of these children and their parents.
2. Material and Methods
We conducted a mixed study with a retrospective part of 3 years (from January 1, 2017 to December 31, 2019) and a prospective cross-sectional period of one month (October 2020). The study involved all children aged 2 to 17 suffering from muscular dystrophy and followed within the framework of a consultation at the Neurological Institute of Simbayah and at the Neurology department of the National Ignace Deen Hospital in Conakry. We proceeded to the identification of the medical files of the patients followed for myopathy. These patients were summoned and re-examined by a neurologist and a neurology intern concomitantly to verify the diagnosis. The diagnosis of myopathy was made by a neurologist certified in pediatric neurology. All children underwent a neuropediatric examination. The diagnostic criteria were essentially clinical based on: a duration of the disease greater than 06 months, an onset beyond 01 years and proximal and distal muscle weakness; often associated with an elevation of CPK and a myogenic tracing on ENMG in accordance with the recommendations of the International Committee for Congenital Myopathies (ICMC) [8] (Iconography 1 and Iconography 2).
2.1. Exclusion Criteria
Presence of one or more of the following: 1) human immunodeficiency virus

Iconography 1. Bilateral calf hypertrophy + bilateral Scapula allata.
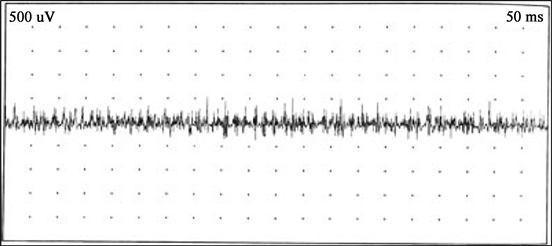
Iconography 2. ENMG trace of an 8-year-old child with gait disorder, calf hypertrophy evolving since birth showing microvolt waves.
(HIV) infection, 2) obstructive hydrocephalus, 3) history of malignancy, 4) evidence of developmental regression, 5) diagnosis of a known genetic syndrome, 6) primary neuromuscular disorder or 7) major extracerebral birth defects. HIV serology was determined by chart review. All children enrolled in the study were HIV negative.
2.2. Data Gathering
We interviewed the mother of each child with a questionnaire including the following variables:
・ socio-demographic data (age, sex, origin, level of education of parents).
・ clinic (reasons for consultation as well as the circumstances of discovery, the severity of motor function disorders was graded according to the Gross Motor Function Classification System (GMFCS) [9]. The type of myopathy distributed according to the international classification of Walton in 1981 [8].
・ At the paraclinical level, all the patients benefited from a dosage of muscle enzymes (CPK, LDH) and an additional biological assessment according to the clinical orientation. ENMG was systematic.
・ The quality of life of children and caregivers was assessed using the Euroqol EQ and PAR-QOL scales [10] [11]. The Euroqol scale includes five items representing five dimensions: mobility, personal autonomy, daily activities, pain and discomfort, anxiety and depression. Each item has five response levels. The answers to these five questions describe the state of health as five consecutive numbers [10].
2.3. Interpretation of PAR-QOL Scores
The scores are transformed into a standardized score by dividing them by the total number of items that compose them. Thus the scores can vary from 1 (no impact at all) to a value of 5 (very impact).
To facilitate the interpretation of the results and to classify the scores by level of quality of life, the scores were grouped in the Grimm-Astruc study [11].
・ Good quality of life: (score less than 2)
・ Average quality of life: (score between 2 and 3.5)
・ Low quality of life (score between 3.6 and 5)
2.4. Statistical Analyses
Statistical analysis was performed using Rstudio software version 1.1456. Descriptive statistics were performed for all variables.
2.5. Ethics
The study was approved and authorization was obtained from the ethics committee of the University Hospital of Conakry. Informed consent was obtained from the parents of each patient.
3. Results
Out of 1195 patients seen in consultation, 17 children presented with a clinical and electrophysiological myogenic syndrome, i.e. a frequency of 1.4%. The average age was 8 ± 4.2 years with extremes of 2 and 17 years. The age group of 6 to 10 years was the most represented with 41.1%. There was a male predominance (64.7%) with a sex ratio M/F = 1.8. An urban origin was found in 64.7%. The average consultation time for our patients was 18.8 months. All patients had motor deficit and axial hypotonia, 10 atrophy (58.8%, 8 gait disturbances (47.2%), 6 hypertrophy (35.2%) (Table 1). Motor deficit according to the GMFCS score 10
![]()
Table 1. Distribution of patients according to socio-demographic and clinical characteristics.
patients were at stage V; 5 at stage IV and 2 at stage III with an average of 4.4 (Table 2). The CPK level was increased in 13 patients, i.e. 76.4% (Mean: 790.5 ± 680.5); 15 (88.2%) patients had elevated LDH levels (mean: 859 ± 667.5). The 17 children had a clinical myogenic syndrome and were confirmed electrophysiologically by the demonstration of a rich and microvolt tracing. All our patients received physiotherapy and 10 (58.8%) corticosteroid therapy combined with vitamin therapy and one (5.88%) received an ACE inhibitor. Descriptive analysis of responses through the PAR QOL scale concluded that myopathy had a medium impact on overall quality of life with a mean score of 2.87 (Table 3) and (Figure 1).
4. Discussion
Our study made it possible to highlight a hospital frequency of CM at 1.4% out of a workforce of 1195 patients. To our knowledge, this is the first study carried out in our context on CM. These pathologies are well described in developed countries with a well-codified international classification [12]. In Africa, very few studies have been carried out on ME given the difficulties of etiological diagnosis and management, the last clinical study dating back 35 years [3]. The limited access to ENMG and the high cost of CPK as well as the absence of a laboratory for the analysis of neuromuscular biopsies explain this scarcity of data. A male predominance was reported in our study which can be justified by the importance that the African community gives to the male sex. The average age of onset is 8.6 ± 4.2 years. A multicenter study conducted in North Africa and Saudi
![]()
Table 2. Distribution of patients according to the GMFCS.
![]()
Table 3. Distribution of patients according to quality of life (Euroquol scale).
![]()
Figure 1. Distribution of parents’ quality of life according to the PAR QOL score.
Arabia reports an average age of between 3 and 5 years. The delay in consultation observed in our study justifies the symptomatology dominated by motor deficits with a GMFS score of V; this could be explained by the ignorance of the neurology department, ignorance and stigmatization of patients with myopathies. This delay in consultation is a predictive sign of seriousness and a factor of poor prognosis overall estimated at 45.31%. The pathologies found were Duchenne muscular dystrophy (n = 20), gamma sarcoglycanopathy (n = 2), infantile spinal muscular atrophy (n = 4), dysferlinopathy (n = 2) and Becker myotonia (n = 1) [5]. Indeed, myology is taught very little, both in basic medical studies and in the specialties of pediatrics and neurology, especially in adults. The diagnostic yield remains low, especially in countries without permanent consultation. This performance can be improved by setting up infrastructures allowing the performance of muscle biopsies and their subsequent analysis. All our patients presented a myogenic tracing to the ENMG; our results can be superimposed on those reported by PJ Lorenzoni et al. [13]. Serum enzyme levels are important in the diagnosis of inflammatory myopathies, and these are generally lower in patients with overlapping myositis than in patients with juvenile dermatomyositis or juvenile polymyositis [13]. The results of our study indicated an increase in serum muscle enzymes in 76.47% of children. The clinico-biological and electrophysiological characteristics made it possible to classify the patients (Table 4). Khadijeh et al. reported a proportion of Duchenne muscular dystrophy (38.9%), girdle and limb myopathy (27%), Becker myopathy (17.2%), facioscapulohumeral myopathy (9.2%) and spinal muscular atrophy (7.7%) [14]. This demonstrates the importance of molecular biology in the diagnosis of MDE.
The management of myopathies is based on a diagnosis established on solid foundations and on an assessment of the prognosis made on a case-by-case basis because these diseases are varied in their clinical presentation and their evolution.
![]()
Table 4. Types of myopathies (clinical-biological classification).
The difficulties of the etiological diagnosis encountered in our context lead us to base the management on the monitoring of symptoms and on the attempt to slow down their evolution and their consequences [15].
Therefore, the choice relies quite heavily on the clinician’s experience in this area. Corticosteroids remain, to date, the first-line treatment for several reasons: their entry into action is one of the fastest among the powerful immunomodulators; they are the best studied therapeutic agents and whose benefits are clearly documented. The rule is initial high-dose corticosteroid therapy (prednisone 1 mg/kg/day). Higher doses showed no benefit, mostly increasing side effects. In our context, quite classically, a good initial response to corticosteroids is observed, but at the same time, corticosteroid-dependent therapy of lesions and relapses affects the long-term prognosis [16].
Beyond corticosteroid therapy, other complementary treatments are necessary given the complexity of neuro-orthopedic, cardiac or respiratory damage.
Currently, there does not seem to be any formal scientific proof of the efficacy of a treatment, whether from a functional or orthopedic point of view [17]. However, technical aids and human resources to preserve physical functions (walking and standing) and quality of life must be put in place as soon as possible in order to make life easier for patients by giving them more autonomy [18] and at the same time ease the burden placed on those around them.
In addition to an analytical rehabilitation focused on the osteoarticular structures to preserve the flexibility of the rib cage (overall flexibility exercises, passive segmental mobilizations, muscle stretching) and on the respiratory muscles to maintain their function (long-term strengthening of the inspiratory muscles , breathing exercises, incentive spirometry), rehabilitation is also based on the early use of mechanical ventilation. In order to maintain the intrinsic qualities of the lung and promote thoraco-pulmonary expansion, these mechanical ventilation supports are largely responsible for increasing the life expectancy of patients with myopathy over the past 20 years [19].
The negative impact of myopathies on the psychological state of patients and their families is not only related to the diagnosis, but also to the fluctuation of signs and symptoms according to the phases [20].
Perceived health measures established by the Euroqol tool are possible within a sample of people with myopathy. In our study, 35.2% of children had mild problems performing routine activities as well as other activities such as bathing and dressing. The parents’ quality of life was moderately emotionally and adaptively degraded with an overall score of 2.87.f.Boyer et al. reported that 30% of parents of myopathic children expressed psychological harm and a clinically significant decrease in quality of life [7].
The ethical and psychological training of health professionals who must support families and patients with myopathies in order to avoid depression seems to be a matter of common sense [8].
5. Conclusion
In our context, the clinical symptomatology of CM was dominated by the complete occurrence of a myogenic syndrome. Beyond the diagnostic problem, the late management of this chronic disabling pathology has a negative impact not only on the quality of life of the child by reducing his autonomy, but also on that of the parents emotionally and adaptively. The establishment of infrastructures allowing the realization of muscular biopsies and their consequent analysis could be an asset for the etiological diagnosis in order to direct a good care.