Microwave Irradiated Palladium-Catalyzed Cascade Type Cross Coupling of Phenols and Halides for the Synthesis of Polyphenolic Ethers ()
1. Introduction
The transformation of simple phenols into a platform of polyphenolic ethereal structure offers an extremely powerful tool in organic synthesis. Phenolic compounds are present in medicinal and edible plants such as; flavonoids, chalcones, coumarins, quinones, phenolic acids [1] - [7]. The antioxidant potential of phenolic compounds shows potent activities for cancer prevention and its treatment. Herein we report a novel route for phenolic ethers based on a cascade type reaction consisting of phenols and trihalo and dihalo compounds. The pioneer works of transition metal catalyzed cross-coupling reactions for C-O bonds done by Buchwald and Hartwig are well cited in the literature. In 1996, Buchwald and coworkers published an article about the synthesis of oxygen heterocycles using palladium catalyst to create C-O bonds [8]. Recently, Buchwald also introduced a new biarylphosphine ligand with [(cinnamyl)PdCl]2 complex for the successful synthesis of diaryl ether (Equation 1) [9].
In 1996, Hartwig and his coworker established a new method for the formation of alkyl aryl ethers in the presence of DPPF-ligated palladium complex to form new C-O bonds [10]. In another article, Hartwig showed the mechanistic studies of the formation of diaryl ether by reductive elimination from the more electron-poor CF3-dppf and Pd-complex. The effect of bulkier ligands is 2 times faster than it was from the dppf complex (Equation 2) [11].
Ma and Chai compared N,N-dimethyl-promoted Cu-catalyzed Ullman type coupling reactions for the synthesis of phenolic ethers from aryl iodides and phenols [12] [13].
Although the palladium catalyzed formation of diaryl ethers is challenging compared to the formation of aryl amines, Buchwald-Hartwig’s outstanding works in this field is a breakthrough. This field does, however, still have ample room to explore. The primary interest of this project was to investigate the possibility of making polyphenolic ethers from phenols and tri- and dihalo compounds under microwave heating. A series of phenols and organic halides have been introduced for this project. One of the palladium complexes, PdCl2(dppf)CH2Cl2, shows significant catalytic effect for the one reaction pot cross coupling cascade process and is able to form polyphenolic ether compounds (Equation 3).
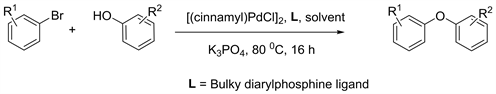
Equation 1. Pd-catalyzed synthesis of diaryl ethers under mild conditions.
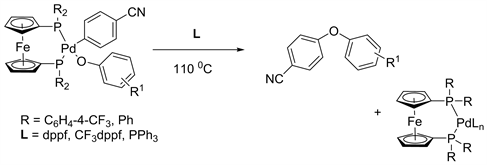
Equation 2. Formation of diaryl ethers from the reactions of dppf-ligated Pd-complex and L.
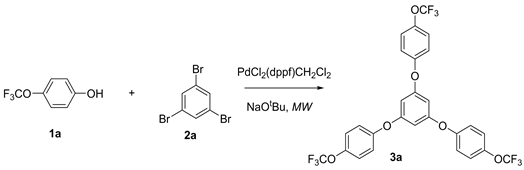
Equation 3. Phenolic ether from 1,3,5-tribromobenzene.
2. Results and Discussion
After running many reactions with different ratios of starting materials, catalysts, solvents, reaction times, temperatures, and bases, several optimized reaction procedures were established. Several palladium complexes such as PdCl2(dppf)CH2Cl2, PdCl2(dtbpf), PdCl2(Ph3P)2, and Pd(OAC)2, were tested. We found that PdCl2(dppf)CH2Cl2 shows effective catalytic effect for this new transformation. Different mole percentages of PdCl2(dppf)CH2Cl2 were applied but most of the results were obtained using 5 mol% of PdCl2(dppf)CH2Cl2. The study showed that excess amount of phenols gave good result when interacting 0.5 mmol of the halides with the load of 5 mole% PdCl2(dppf)CH2Cl2. The results are summarized in Figure 1. All of the products shown in Figure 1 are solvent free systems. Several solvent systems such as, 1,4-dioxane, isopropanol/water, toluene, THF, CH2Cl2, and acetonitrile, were applied and failed. In the control experiment, the predicted cross coupling product 3a was totally undetected in the absence of palladium catalyst. The experiments worked well when microwaved at 80˚C for 3 - 4 h. In the case of sp3 1,3-dibromopropane 2b, reactions microwaved for 3 h (Obs. 2, 3, and 4 in Figure 1).
![]()
Figure 1. Solvent free Pd-catalyzed cross-coupling of phenols and halidesa. aAll yields are isolated yields and purified by colum chromatography or preparative TLC. 5 mole% PdCl2(dppf)CH2Cl2 were used as catalyst. No solvent.
These reactions when run with equimolar amount of NaOtBu, no product was found but in presence of 5 mole% palladium catalyst similar reaction showed cross-coupling product. In case of 1,4-diiodo butane, smooth reaction was observed with excess amount of NaOtBu without catalyst. It is possible that although diiodo sp3 alkyl halide works well without palladium catalyst in simple SN2 reaction for phenolic ether synthesis, relatively slower dibromo sp3 alkyl halide such as 2b possibly promoted the reaction by insertion of palladium and favors cross-coupling phenolic ether compounds.
When the same reaction was run without palladium catalyst, no change was observed except for two clear spots of starting materials in TLC. This reaction is also futile to produce cross-coupling product in the absence of solvent even with a large excess of phenol, which usually worked in other cases shown in Figure 1. The product 3e was obtained in moderate yield. Under the same reaction conditions, 1-bromo-3-iodobromobenzene 2c furnished good results with other phenols (Obs. 2, 3, 4 Figure 2). Instead of 1-bromo-3-iodobromobenzene 2c, these reactions were run with 1-chloro-3-iodobenzene as well. 2c yielded better coupling products.
![]()
Figure 2. Pd-catalyzed cross-coupling of phenols and halides with solventa. aReactions microwaved for 5 h at 140˚C in presence of IPA/water (2:1) solvent system. 2.5 mole% PdCl2(dppf)CH2Cl2 used as catalyst. b2.0 mmol (195 mg) of NaOtBu used.
The cross coupling of bromo iodomethane 2d gave the desired product when interacted with phenol 1c in the presence of 1,4-dioxane as a solvent. To furnish this phenolic ether product, 1.1 mmol (103.5 mg) of phenol, 0.5 mmol of bromo iodomethane, 5 mol% of PdCl2(dppf)CH2Cl2, and 2 mmol of NaOtBu were used. The reaction was in the microwave oven for 5 h at 80˚C (Equation 4).
Interestingly, we attempted a mixture of phenols and halides in the presence of palladium catalyst to see the scope of this new transformation to phenolic ether.
3. Experimental Procedure
Procedure 1. This procedure is a representative one. In an oven dry, clean microwave vial loaded with 2.0 mmol of NaOtBu (195 mg), 5 mol% of PdCl2(dppf)CH2Cl2 (22.0 mg), was capped with an air-tight silicon septum, The reaction vial was flushed with argon followed by the addition of 0.5 mmol of the 1,3,5-tribromo benzene (160.0 mg) via micro syringe and an excess amount (1.5 mL) of 4-trifluoromethoxy phenol via dry syringe. The resulting reaction mixture was microwaved at 80˚C for 3.5 h. The crude reaction product was subjected to column chromatography with hexane and ethyl acetate (25:1) as eluents and the polyphenolic ether product was collected. The slurry of 40 mL (by volume) weight of silica gel and eluent (Hexane:ethyl acetate 25:1) was transferred into the column, tapped to remove air, and packed. The column was ther ready to use for separation. 10 g of silica gel were added to the crude reaction product. After rotary evaporation, we got a fine powder, which was transferred to the surface of the column. A layer of sand was added above it to make sure that the powder layer was unbroken. All collected fractions were monitored by thin layer chromatography to identify the new spots for desired phenolic ether compound. We collected all those fractions with same Rf value in a clean and dry round bottom flask. The solvent with product was evaporated using the rotary evaporator, and then was added dichloromethane to get rid of any excess of ethyl acetate that could remain in the product. The product was dried under the vacuum pump for several hours to make sure that the solvent did not remain in the product. Finally, the concentrated product was analyzed by 1H, 13C, and 19F NMR spectroscopy. Compound 3a. 1H NMR (CDCl3, 400 MHz) δ 7.12 (m, 7H,), 6.83 (m, 8H). 13C NMR (CDCl3, 100 MHz) δ 153.9, 142.9, 124.3, 122.6, 121.8, 119.2, 116.4. 19F NMR (CDCl3 400 MHz) δ −58.4.
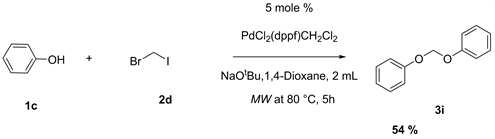
Equation 4. Cross coupling of bromoiodomethane and phenol.
Procedure 2. The formation of ether 3e by cross coupling reaction involved 1.65 mmol (155.28 mg) of phenol 1c and 0.5 mmol (160.6 mg) of 1,3,5-tribromobenzene 2a. The reactants were loaded in dry, clean microwave vial and 2.65 mmol (255 mg) of NaOtBu and 2.5 mol % of PdCl2(dppf)CH2Cl2 were added to the mixture. Then it was capped with septum and flushed with argon followed by the addition of iso-propanol/water (2:1) as solvents (Obs. 1, Figure 2). The resulting mixture irradiated at 140˚C for 5 h. The crude reaction mixture was filtered through a sintered funnel and concentrate. For purification, the crude mixture was subjected to column chromatography using hexane/ethyl acetate (25/1) as eluents.
Procedure 3. An oven dry, clean microwave vial was loaded with 2.0 mmol of NaOtBu (195 mg), 5 mol% of PdCl2(dppf)CH2Cl2 (22.0 mg) and capped with air-tight silicon septum. The reaction vial was flushed with argon followed by the addition of 0.5 mmol of the bromo iodomethane 2d via micro syringe. 0.5 mmol (48.0 mg) of phenol 1c and 0.5 mmol of 4-trifluormethoxy phenol (64 μL) 1a were added via micro syringe as well. The resulting reaction mixture was flushed in argon then was added the solvent 1,4-dioxane (2 mL) The loaded reaction vial was microwaved at 80˚C for 5 h. The crude reaction product in the reaction tube diluted with ethyl acetate was transferred into a separatory funnel. After a standard extraction with brine solution, the organic layer was collected in a small Erlenmeyer flask over anhydrous Na2SO4. The ethyl acetate layer was filtered through a sintered funnel and the collected filtrate in a round bottom flask was completely dried by rotary evaporator in vacuo. The column chromatography technique was used to sperate the product. The desired polyphenolic ether product 3j was confirmed by 1H NMR, 13C NMR, and 19F NMR (Equation 5).
4. Probable Reaction Mechanism
The probable mechanism of this palladium-catalyzed polyphenolic ether reaction most likely proceeds via a pathway shown in Figure 3. The oxidative addition of the Pd(0)Ln with the trihalide renders the Pd(II) inserted organometallic intermediate. In the presence of sodium tertiary butoxide, the chelation/deprotonation of phenol renders the species that is ready to do reductive elimination to yield the first ether moiety. It is rational to predict that the remaining halides follow the same catalytic process with regenerated palladium catalyst and furnish the carbon-oxygen bonds for polyphenolic ether product.
![]()
Equation 5. Phenolic ether from mixed phenols.
![]()
Figure 3. Probable reaction mecanism of polyphenyl ether.
5. Conclusions
This work developed a microwave irradiated new synthetic processes for the synthesis of a good number of polyphenolic ethers from phenols and aromatic tribromo- and 3-iodo-bromobenzene in one reaction pot. The cross coupling of phenols and alkyl halides such as 2b and 2d also successfully steered in single reaction vial. The multiple carbon-oxygen bonds formation reaction is one pot reaction is not known through metal-catalyzed cross-coupling reaction. Since antioxidant nature polyphenolic ether compounds are potent activities for cancer treatment, the new synthetic method of multi coupling and the making of multiple carbon-oxygen bonds in one reaction vial will get much attention among chemists. The initial findings of making phenolic ether from the two different phenols and halides (Equation 5), will be explored and reported in due courses from our laboratory.
NMR data of products from Figure 1, Figure 2, 3h and 3i are given below.
Compound 3a, 1H NMR (CDCl3, 400 MHz) δ 7.12 - 6.83 (m, 15H). 13C NMR (CDCl3, 100 MHz) δ 153.9, 142.9, 124.3, 122.6, 121.8, 119.2, 116.4. 19F NMR (CDCl3 400 MHz) δ −58.4;
Compound 3b, 1H NMR (CDCl3, 400 MHz) δ 7.27 - 6.88 (m, 8H), 4.15 (m, 4H), 2.27 (m, 2H). 13C NMR (CDCl3, 100 MHz) 157.3, 142.7, 122.4, 119.4, 115.1, 64.6, 29.1 19F NMR (CDCl3 400 MHz) δ −58.4;
Compound 3c, 1H NMR (CDCl3, 400 MHz) δ 7.75 - 7.65 (m, 8H), 4.43 - 3.61 (m, 2H), 4.28 - 4.0 (m, 2H), 2.21 (m, 2H). 13C NMR (CDCl3, 100 MHz) δ 168.6, 163.5, 134.4, 133.9, 132.8, 128.8, 123.3, 60.2, 31.3, 29.2, 20.7, 14.0;
Compound 3d, 1H NMR (CDCl3, 400 MHz) δ 7.33 - 6.93 (m, 10H), 4.19 (m, 4H), 2.29 (m, 2H). 13C NMR (CDCl3, 100 MHz) δ 1158.9, 129.4, 120.7, 114.5, 64.3, 29.3;
Compound 3e, 1H NMR (CDCl3, 400 MHz) δ 7.74 - 7.1 (m, 15H, 3H) 13C NMR (CDCl3, 100 MHz) δ 141.8, 141.2, 129.9, 129.1, 128.8, 127.4, 127.2, 126.1;
Compound 3f, 1H NMR (CDCl3, 400 MHz) δ 8.31 - 6.41 (m, 12H, Aromatic). 13C NMR (CDCl3, 100 MHz) δ 155.8, 1417, 128.6, 127.1, 124.3, 122.2, 121.8, 119. 2, 116.2, 19F NMR (CDCl3 400 MHz) δ −58.7;
Compound 3g, 1H NMR (CDCl3, 400 MHz) δ 8.31 - 6.41 (m, 12H, Aromatic). 13C NMR (CDCl3, 100 MHz) δ 161.7, 139.7, 135.4, 131.3, 130.8, 129.8, 125.7, 123.0, 118.9, 117.4, 112.9, 94.4. 19F NMR (CDCl3 400 MHz) δ −61.6;
Compound 3h, 1H NMR (CDCl3, 400 MHz) δ 7.05 - 6.68 (m 14H, Aromatic). 13C NMR (CDCl3, 100 MHz) δ 155.5, 128.6, 119.2, 114.6;
Compound 3i, 1H NMR (CDCl3, 400 MHz) δ 7.23 - 6.82 (m 10H, Aromatic), 6.03 (s, 2H). 13C NMR (CDCl3, 100 MHz) δ 155.6, 129.5, 120.5, 116.4, 115.3;
Compound 3j, 1H NMR (CDCl3, 400 MHz) δ 7.60 - 6.82 (m, 10H), 7.12 (s, 2H). 13C NMR (CDCl3, 100 MHz) δ 156.0, 155.0, 142.3, 129.5, 122.4, 120.3, 116.1, 115.3. 19F NMR (CDCl3 400 MHz) δ −58.4.