Bone marrow increases human islets insulin positive cells in co-culture: quantification with flow cytometry ()
1. INTRODUCTION
Diabetes is a condition whereby the body is not able to regulate levels of glucose (a sugar) in the blood, resulting in too much glucose being present in the blood. Pancreatic islet transplantation is a promising treatment for patients with Type 1 diabetes mellitus, which results in hyperglycemia due to islet β cells damage [1-4] . However, maintaining the structural integrity and insulin release of post-isolated human islets is a pre-requisite for achieving successful islet transplantation. Existing protocols for maintaining human islets in vitro are limited, allowing only short-term (less than 72 h) storage of cells under conventional static culture conditions [5]. Extending the duration of culture beyond this period usually results in a significant loss of structural integrity within the islets and a decline in glucose-stimulated insulin release [6-8].
Our previous studies have indicated that bone marrow can maintain β cell function by evaluating insulin secretion even after 200 days of in vitro culture [9]. Exploration of mechanisms how BM support human islets in long term suggests that BM cultured human islet may increase β-cell population in islet during culture. However, quantification of β-cell from cultured human islet is a challenge. There are several methods to identify β-cells, including methods to determine the function and purity of islets. At the mRNA level, polymerase chain reaction (PCR) and in situ hybridization are performed. Enzymelinked immunosorbent assay (ELISA) is used to measure insulin secretion [10-12] or c-peptide release [13] in supernatants. However, ELISA measures only β cell insulin secretion over time, but not the number of insulinproducing cells within this heterogeneous cell population. For insulin producing cell numbers, insulin-specific immunohistochemistry combined with light microscopy [14, 15] can be used. This method has its limitations in terms of reliability and comprehensiveness. Another method is the use of flow cytometry on single cell suspensions with fluorescent zinc probes [16,17] . This method uses FluoZin-3 staining to reveal higher amounts of chelatable zinc ions in β-cells [18]. However, we did not successfully detect positive FluoZin-3 in dissociated monolayer islet cells in long term culture in vitro because this method also stained some kind bone marrow cell like monocyte [19] and T cell [20]. So this method will result false positive β-cells in islet and bone marrow co-culture. Other methods include the use of cell surface molecule Poly-Sialated Neural Cell Adhesion Molecule (PSA-NCAM) as a marker of β cells, which is also suitable for flow cytometry analysis and cell sorting [21] . However, PSA-NCAM lacks specificity since it expresses in different tissues such as nerve cells [22,23] . In this study, a two-step protocol using intracellular insulin staining and flow cytometry was established in our group and applied to quantify insulin positive cells from a culture containing a heterogeneous mixture of islet cells. Using this technique, we were able to quantitate insulin-producing β cells from culture islets with or without BM. Our studies support the hypothesis that BM supporting human islet β cell function includes increase β cell population while not in islet only culture.
2. MATERIALS AND METHODS
2.1. Antibodies and Reagents
4% paraformaldehyde (Sigma) solution was prepared by dissolving paraformaldehyde powder in phosphate buffered saline (PBS, Invitrogen, Carlsbad, CA, USA). Insulin antibody (ab7842) and insulin + proinsulin antibody (ab20756) were purchased from Abcam (Cambridge, MA, USA). Proinsulin antibody (10-P14B) was purchased from Fitzgerald Industries (Acton, MA, USA). Normal IgG of goat (sc-2028), rabbit (sc-2027), mouse (sc-2025) and guinea pig (SC-2711) were purchased from Santa Cruz biotechnology Inc (Santa Cruz, CA, USA). Fluorescein isothiocyanate (FITC) conjugated goat polyclonal to Guinea pig secondary antibody (ab6904) was purchased from Abcam. FITC conjugated horse antimouse secondary antibody (FI-2000) was purchased from Vector Laboratories (Burlingame, CA, USA).
2.2. Tissue and Cell Culture
Human pancreatic islets. Human islet tissue from normal donors was obtained through the Islet Resource Centers (ICRs) Basic Science Islet Distribution Program from the Human Islet Laboratories at the University of Pennsylvania (Philadelphia, PA, USA), Massachusetts General Hospital (Boston, MA, USA), City of Hope National Medical Center (Duarte, CA, USA), University of Miami, School of Medicine, Diabetes Research Institute (Miami, FL, USA). The use of these cells is approved by the Institutional Review Board (IRB) at Roger Williams Hospital and the ICR Committee [9].
Human BM. Human BM from normal donors was obtained under a separate Roger Williams Hospital IRBapproved protocol. Bone marrow erythrocytes were eliminated by Ficoll-Paque™ Plus (Amersham Biosciences; Amersham, UK) per manufacturer directions. Cells were then washed twice with 5% Fetal Bovine Serum (FBS) in phosphate buffered saline (PBS) and resuspended in culture medium (see below). Trypan blue (Gibco, 15250-061) staining was used to assess cell viability. The use of these cells is approved by the Institutional Review Board (IRB) at Roger Williams Hospital and the ICR Committee [9].
Allogeneic BM co-culture with human islets. Human islets were received from ICR within 48 h of harvest from cadaveric donors. The viability of whole islets upon arrival was >95% as determined by trypan blue dye exclusion. Islets were placed in culture at 50 islet equivalents (IEQs) per ml with 1 × 106 allogeneic BM cells/ ml. Cultures were maintained in RPMI 1640 (Gibco) supplemented with 10% heat inactived FBS (Hi-FBS, Hyclone), 5.5 mM glucose, 10 mM HEPES, and 1% P/S, pH 7.3 [9].
2.3. Enzyme-Linked Immunosorbent Assay for Insulin Measurement
Insulin concentrations in each specimen (culture medium and cell extracts) were measured using Ultra Sensitive Human Insulin ELISA Kit (Millipore, EZHI-14K) according to the vendor’s instructions. Insulin concentrations were measured and calculated using a μQuant microplate reader equipped with KC Junior® microplate reader software (Bio-Tek Instruments, Inc., Winooski, VT) [24] . Briefly, insulin standards and appropriately diluted samples were added to 96-well microplates precoated by the manufacturer with an anti-insulin antibody and incubated for 1 hour at room temperature. Samples were removed, plate was washed five times, and the enzyme-linked anti-human insulin conjugate was added to the well for 30 minutes incubation at room temperature. After washing five times, the enzyme substrate solution was added and incubated 10 to 15 minutes at room temeperature in the dark. The reaction was halted by adding 1 N sulfuric acid. Absorbance at 450 nm was read with a μQuant microplate reader and insulin concentrations were calculated based on the standard curve by KC Junior® microplate reader software (Bio-Tek Instruments, Inc.) [9].
2.4. Two-Step Intracellular Anti-Insulin Staining and Flow Cytometry
After human islets were dissociated into single cells with 0.05% trypsin-EDTA and 0.2% collagenase, cell suspensions were washed and then re-suspended in PBS for cell counting. The cell viability was measured with trypan blue. Cells were fixed in 2% paraformaldehyde solution for 20 min in the dark at 4˚C with slight shaking every 5 minutes. Cells were then washed in ice-cold 5% Hi-FBS PBS and permeabilized in 0.05% triton x-100 solution in 5% Hi-FBC PBS for 15 minutes at room temperature. Cells were washed and resuspended with icecold PBS buffer (including 0.05% triton x-100 and 5% Hi-FBS). Cell suspensions were added to each roundbottom polystyrene tube for the antibody staining. Cells were stained with isotype control or antibody to insulin or proinsulin and an antibody that recognizes both proinsulin+insulin at 1:50, 1:100 and 1:200 dilution diluted in 0.05% triton x-100 and 5% Hi-FBS PBS for 1 hour in ice or overnight at 4˚C. After incubation, cells were washed and resuspended in ice-cold permeablization buffer. Cells were then stained with the secondary antibody conjugated with fluorescein isothiocyanate (FITC) at 1:300 (FITC conjugated goat polyclonal to Guinea pig secondary antibody) or 1:100 (FITC conjugated horse antimouse secondary antibody) dilution diluted in ice-cold permeablization buffer for 30 minutes on ice and in the dark. For the non-specific staining, cells were only stained with specific secondary antibody conjugated with FITC for 30 minutes on ice and in the dark. Finally, cells were washed in permeablization buffer by centrifugation at 300 g for 10 minutes and resuspended in 400 μL 5% Hi-FBS PBS buffer. Flow cytometry analysis was performed on Becton Dickinson LSRII system (BD Biosciences, CA, USA). In all cases, more than 10,000 cellular events were analyzed per tube, and in several cases, 100,000 cellular events were analyzed per tube. This method is relative stable than other reported methods [25] .
2.5. Data Analysis
Results are displayed as mean ± standard deviation (SD). Statistical significance was determined using the Student’s t test; p values less than 0.05 are considered statistically significant.
3. RESULTS
3.1. Optimizing Antibodies for Labeling β-Cell
Cultured human islets are cluster of cell group, which have to be dissociated into single cells. We established a protocol to dissociate these cultured human islet into single cells. Human islets were dissociated under the optimized digestive conditions (0.05% trypsin and 0.2% collagenase solution) into single cells (Figure 1). To find an optimized antibody and concentration for labeling β cells, cells were stained with various concentrations of antibodies against insulin, proinsulin, or insulin + proinsulin. Optimum staining for insulin occurs at a 1:100 dilution of anti-insulin antibody, at 1:200 of anti-proinsulin antibody, and 1:100 dilution of anti-insulin + proinsulin antibody. Under same condition of islet dissociation samples, results showed that anti-insulin antibodies identified the least amount of cells (17.2% ± 0.44%) whereas anti-proinsulin detected 45.4% ± 3.48% cells and the antibody for insulin + proinsulin detected 43.2% ± 7.92% cells. The antibodies that recognized either proinsulin or proinsulin + insulin were two folds greater versus insulin only antibody within similar groups (Figure 2), then we used anti-proinsulin antibody in following studies.
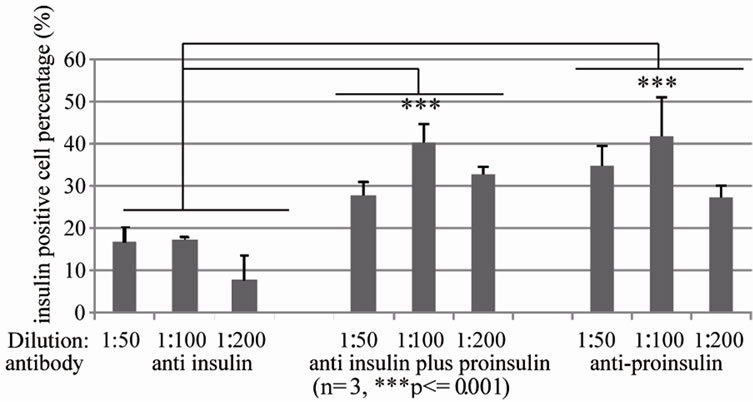
Figure 2. Optimized anti-insulin antibodies for identifying human islet β cells dissociated and recovered human islet cells were fixed and stained with anti insulin antibody (left), anti insulin plus proinsulin antibody (middle) and anti-proinsulin antibody (right) at 1:50, 1:100, 1:200 dilution respectively, and then subjected to flow cytometry analysis (n = 3, ***p ≤ 0.001).
3.2. Quantification of Human Islet β Cells with or without BM from Different Culture Days
We evaluated β cell populations in human islets with or without BM during culture from day 3 to 45 as indicated by insulin positive cells with the established β-cell quantification method. We demonstrated that the β-cell population decreases in islet only cultures from 16.8% ± 0.8% on Day 3 to 7.9% ± 1.9% on Day 10 and then to 0.93% ± 0.06% on Day 45 (Figure 3(a)).Consequently, insulin release decreased from 19412.4 ± 104.3 µU/ml on Day 3 to 11386.7 ± 5920 µU/ml on Day 10 and to 418.0 ± 134.5 µU/ml on Day 45 (Figure 3(b)).
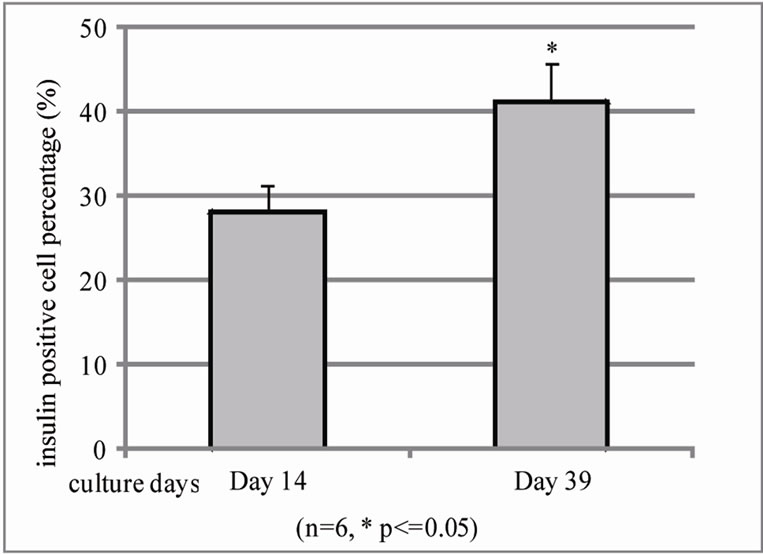
In evaluation of BM co-culture human islet during different culture days using same method, we found that β cell population from islet with BM co-culture significantly increases from 14 to 39 day in cultures (28.15% ± 3.4% on day 14 and 42.3% ± 4.5% on day 39 cultures) (Figure 4(a)) and function evaluation also found insulin release increase but too much variation to statistic sig-
 (a)
(a)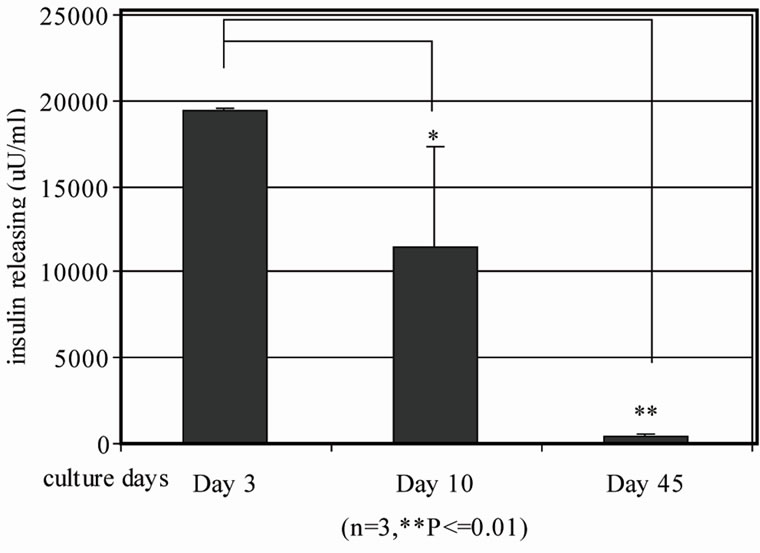 (b)
(b)
Figure 3. Quantification of insulin positive cells and insulin releasing in human islets during different culture days by flow cytometry and human insulin ELISA. Human islets were cultured for 3 days, 10 days and 45 days in vitro respectively. (a) Human islets were then dissociated with 0.2% collagenase + 0.05% trypsin into single cells. Guinea pig anti-human insulin polyclonal antibody and FITC conjugated secondary antibody were subsequently added to stain insulin positive cells and then subjected to BD LSRII flow cytometry analysis (n = 3, *p ≤ 0.05, **p < 0.01,); (b) Human islet insulin release was analyzed by human insulin ELISA kit.
nificance (4650 ± 866.51 µU/ml on day 14 and 6748.68 ± 2236.59 µU/ml, Figure 4(b)).
Comparison between islet with or without BM in different culture days, we found that there are significantly variation during two weeks and short period cultures, islet only groups (16.5% ± 9.19) and islet and BM coculture groups (28.15% ± 3.4%) (Figure 5(a)). However, insulin positive cells in islet with BM co-culture on Day 39 was significant increase 42.3% ± 4.5% versus 1.15% ± 0.78% for islet only cultures. The function of β-cell evaluation indicated that short term cultured human islet only insulin release is higher than BM co-cultured islet but not significance because variation (Figure 5(b)). We suspect that this increasing insulin release may resultfrom huge amount of β cell death in culture. The signifi-
 (a)
(a)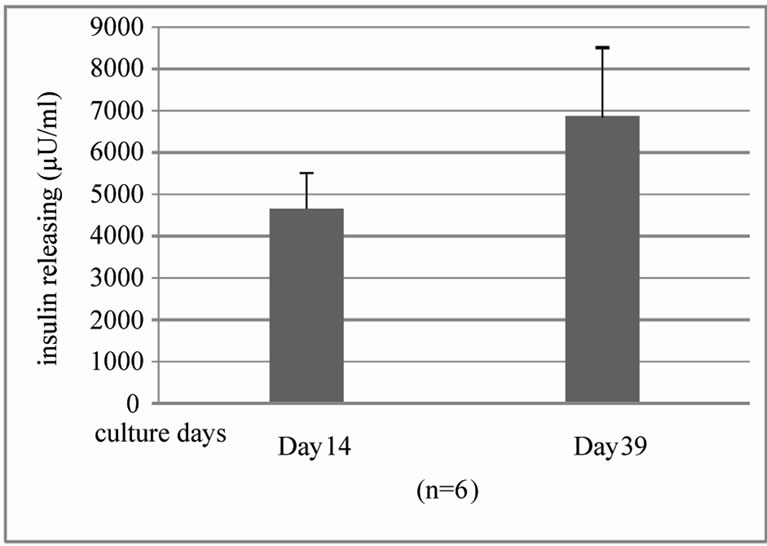 (b)
(b)
Figure 4. Quantification of insulin positive cells and insulin releasing in islet-BM co-culture between day 14 and 39 by flow cytometry and human insulin ELISA. (a). Dissociated and recovered human islet cells were fixed and stained with anti insulin antibody. Insulin positive cells were detected with flow cytometry analysis on the 14th day (left) and 39th day (right) of human islet culture (n = 3, *P < 0.05). B. Insulin release was evaluated by human insulin ELISA kit.
cant reduction of β-cell number and insulin release in continuing culture supports this notion (Figure 5).
4. DISCUSSION
Several recent studies have suggested that adult bone marrow cells can influence beta-cell regeneration in diabetic animals [26-32]. Other reports, however, have contradicted these findings [33,34]. In this study, we addressed this question by establishing a method to quantify insulin positive cells in human islet cultured with or without bone marrow. The optimized islet culture dissociation conditions coupled with methods to label single insulin positive β cells described here allows us to quantify cultured β-cells from islet clusters.
In terms of antibodies for detecting insulin positive cells, we expected that an antibody capable of detecting both insulin and proinsulin would yield the highest sensitivity due to its wide range of binding capability for proinsulin and insulin and the results support this notion that proinsulin-only and insulin + proinsulin antibodies showed to have better results than insulin only antibody. Within the β cell granules, proinsulin is converted by a process of enzymatic cleavage to insulin and C-peptide [35,36] but over 90% of the newly generated insulin is released and only 0.5% of proinsulin is rapidly released [37]. Given this, using proinsulin antibodies for labeling β cells is optimal for flow cytometry quantification.
Adult bone marrow harbors cells to have pluripotent differentiation capacity and benefit other tissue regeneration. Pluripotent mesenchymal stem cells in adult bone marrow have the capacity to differentiate into a number of neuroectodermal [38], endothelial [39] , epithelial [40] , and muscle [41] cells. Bone marrow is capable of maintaining pancreatic islets in the absence of immunosuppression, and can constitute an immunoprivileged environment for engraftment [31,42] . Transplantation of bone marrow derived cells into diabetic mice can reducehyperglycemia through proliferation of β-cells, differentiation of pancreatic stem cells to contribute to the rege-
neration of β-cells [28,29,43,44] , and improve islet graft function by promoting graft vascularization [31,45] . Bone marrow’s ability to correct hyperglycemia in vivo has been shown in cases where animals with islets transplanted into bone marrow had a higher chance to reach euglycemia than islets transplanted in the liver [46]. With the hypothesis that bone marrow supporting human islet in vitro includes increase β cells, we used our established quantification method to evaluate effects of bone marrow on the quantity of human islet β cells during culture with or without bone marrow. We found that bone marrow increases the number of insulin positive human islet β cells during long term culture on the 39th day vs the 14th day (Figure 4) and significantly increase β cell function vs. islet only in long term culture (Figure 5). We still have no answer why bone marrow cultured human islet taken so long (two or three weeks) to see the significant increase of β cell population but it may be involved bone marrow repair islet injury and islet recovery from isolation shock process which need time to recover. Islet only without bone marrow support is in slow process to loss.
In summary, we have demonstrated that human islet cultured with BM significantly increase β cells function which partially resulted from β cell population increase. Newly established method with two steps of labeling β cells coupled with flow cytometry to quantify cell samples (≥30,000 cells) in a fully objective manner is repeatable with little expense. The results indicated that BM enables supporting human islet culture by increase insulin positive human islet β cells while islet only culture lost β cells in same culture period. Further studies may be required to determine the mechanisms how bone marrow contributes to increase β cells in human islet culture.
5. CONCLUSIONS
We have demonstrated that BM supporting human islet survival and function in long term culture. It includes increase human islet β cell population evaluated by a flow cytometry-based analysis method to quick and accurate quantification of insulin-positive islet β cells from cultured human islets. Further studies for the mechanisms involved will be explored in future.
6. COMPETING INTERESTS
The authors have declared that no competing interests exist.
7. ACKNOWLEDGEMENTS
This work was funded by NIH P20RR018757 (COBRE, PI Dr. Falanga), JDRF 1-2007-180 and Roger Williams Hospital Research fund for Dr. Luo’s Research. We thank the Islet Cell Resource Centers (ICR) funded by the NIH and JDRF for distributing human islets for this project, Cell Sorting and Image Core was funded by NIH/NCRR 5P20RR018757, Dr. Camillo Ricordi’ valuable advises.