The chemical mechanism of oxidative stress due to the non-transferrin-bound iron (NTBI) ()
1. INTRODUCTION
It is generally recognized that iron, the most abundant transition metal ions in mammalian systems, is a necessary trace element and is required for normal metabolic processes spanning molecular oxygen transport, respiratory electron transfer, DNA synthesis, and drug metabolism [1,2]. In fact, iron deficiency leads to the deficiency of neurotransmitters such as dopamine and serotonin in brain, inducing several mental diseases such as Parkinson’s disease, depression and schizophrenia, etc. [3] Thus, the ancient Greeks concocted potions of iron filings dissolved in vinegar, hoping that drinking this liquor would empower them with the properties of the element [1]. Accordingly, for several decades we have sold the idea that “iron-fortified” foods will make us healthier and stronger, when in truth, this holds only for the persons who are frankly iron deficient. For the majority of persons, iron supplementation simple results in ever-increasing store of excess iron in body.
In human body, we have no real mechanism for the elimination of excess iron, and as a result,cells continuously store excess absorbed iron in a complex with the protein ferritin. Normal cells store iron mainly in ferritin molecules [4,5], but under conditions of iron excess some of it is shunted into another storage form known as hemosiderin, in which the excess iron is deposited as polymeric iron(III) with oxo-bridges. Hemosiderin is typically insoluble. Plasma iron is normally bound to the iron transport protein transferrin. When some chelates (amino acids derivatives, small peptides or citrate, etc.) are present in the plasma, the hemosiderin and iron deposition which contains polymeric iron(III) with oxobridges may dissolve; these water-soluble iron ions not associated with transferrin is generally termed as nontransferrin-bound iron (NTBI). NTBI is detected in the plasma of patients with thalasseimia, hemochromatosis and other iron-overloading disorders, and is present at concentration up to 10 μM [6-8]. It should be noted here that NTBI has been thought to play an important role in iron induced cell damage with resultant peroxidation of cell membrane lipids and other biomolecules, and such oxidative damage is implicated as an important contributor in the pathogenesis of cancer, cardiovascular disease, aging and other degenerative diseases.
Despite numerous studies over the last 30 years since plasma NTBI was first postulated to exist, it is still poorly characterized. At present, one of the most definitely definable NTBI should be iron deposition, which is frequently observed for several organs of patients of hemochromatosis, other iron-overloading disorders, and acerupulasminemia, which will be discussed in Sections 3 and 4. In this review, we will demonstrate the several chemical models for NTBI, and show the chemical origin of iron toxicity in human body due to NTBI on the basis of my new concept on the mechanism of oxygen activation in biological oxygenases [3,9,10], and propose the new technique to eliminate the NTBI with safety.
2. STRUCTURAL PROPERTY OF ACTIVE SPECIES TO INDUCE RENAL PROXIMAL TUBULAR INJURIES AND RENAL CARCINOMA BY ARTIFICIAL IRON(III) CHELATES
2.1. Structural and Chemical Properties of Iron(III)-nta Chelate and Its Peroxide Adduct
Ferric nitrolotriacetate (Fe(III)-nta) is a well-known renal carcinogen, and Fe(III)-nta-injected animals have been used as a model of carcinogenesis [11-13]. When Fe(III)- nta is intraperitoneally injected into animals, lipid peroxidation and oxidative modification of proteins and DNA occur in renal proximal tubules, and tubular epithelial cells are damaged. Thiobarbituric acid reactive substance (TBARS [14]; these include malondialdehyde and other aldehyde derivatives) has also been shown to increase in kidneys, and cold Schiff staining showed lipid peroxidation in renal proximal tubules in Fe(III)-ntatreated animals. In Fe(III)-nta-injected mice, amount of reduced glutathione decreased, and oxidixed form increases when metabolic rate of glutathione was accelerated, and repeated injections of with Fe(III)-nta result in appearance of atypical epithelial cells in renal tubules, and finally in induction of renal carcinoma. Kawabata et al. reported that some damaged tubular cells disappeared from the tubules due to apoptosis in Fe(III)-nta injected mice [12].
From all these studies, there is no doubt that Fe(III)- nta induces renal carcinoma, but detailed molecular mechanism of Fe(III)-nta induced carcinogenesis, i.e., chemical properties responsible for renal tubular injury and carcinogenesis, remain unknown at present. In order to answer the above questions, which at the same time will contribute to clarify the chemical composition of NTBI and the origin of the toxicity due to NTBI, we have continued to study on the chemical mechanism of renal injury and carcinogenesis by many iron(III) chelates analogous to (nta) [13], as described in Figure 1.
As the results, we have found that the proximal tubules necrosis induced by artificial iron(III)-chelates in rat kidneys are highly dependent on the chelate structure (see Table 1 and Figure 2), and injuries such as lipid peroxidation and protein oxidation are observed mainly in the renal proximal tubules, but any injuries were not observed in the distal position [13]. It should be noted here that not all the iron(III) ions in plasma are toxic! The origin for the above facts was elucidated on the basis of my new concept on the mechanism of oxygen activation in biological oxygenases [3,11,12].
We have determined that crystal structures of several iron(III) compounds including (nta) [15,16]. As shown in
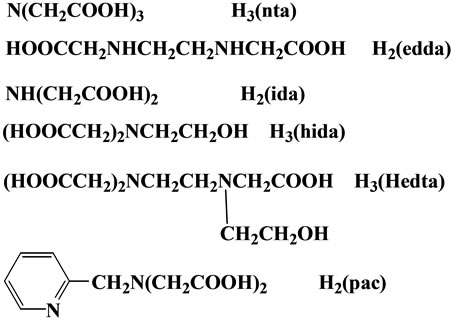
Figure 1. Several chelates cited in this review.

Table 1. Effects of Iron Chelates on renal tubular injuries [13]. (+, active; –, inactive).

Figure 2. Proximal tubules necrosis in rat kidneys 6 h after injection of (A) Fe(III)-(ida) at pH 6.2; (B) Fe(III)-(ida) at pH 8.2. Hematoxylinand eosin staining (magnification, 100). Proximal tubular cells were specifically destroyed by Fe(III)-(ida) at pH 5.2, 6.2, and 7.2. Effects of Fe(III)-(ida) at pH 8.2 were not observed. Patchy degeneration of the proximal tubular epithelium with pyknotic nuclei (small arrowheads). Regenerative cells are large and irregularly shaped with prominent nucleoli (large arrowheads).
Figure 3, Fe(III)-(nta) complex is of a dimeric structure with oxoand carbonato-bridge. The bent Fe-O-Fe core is consistent with the unique absorption spectra [16]. The structure of the Fe(III)-(edda) complex should be similar to that of the (nta)-chelate based on the absorption spectrum of this compound. It should be noted here that although the crystal structure of the Fe(III)-(pac) complex is of a dimeric structure similar to that of the (nta)-compound, tabular injury by the Fe(III)-(pac) compound is negligible (see Table 1) and the Fe(III)-(pac) compound does not induce the renal caricinoma, and that tabular injuries by the Fe(III)-(edda) compound is much less than that by the (nta)-complex (see Table 1) [13,16].
When we compared the chemical properties of the three iron(III) compounds including (nta), (edda) and (pac) ligands, notable differences were detected; at first, the activity for TBARS formation in the presence of ribose and hydrogen peroxide is remarkable for the Fe(III)-(nta) complex, but the effect by the Fe(III)-(pac) compound is negligible, and that by the Fe(III)-(edda) is the much weaker than that of the (nta)-chelate [16].
Crystal structural data of the two compounds, (nta) and (pac) have revealed that the dimeric structure of the (pac) complex is more stronger than that of the corresponding (nta)-compound, because the distances both the Fe-O (oxo oxygen) and Fe-O (carbonato ion) distances are shorter in the (pac)-compound than those in the (nta)-compound [16]. Based on these facts including absorption and ESR spectral data, it seems quite reasonable to conclude that the active species to decompose ribose to give TBARS in the presence of hydrogen peroxide should be a (μ-η1:η1)-peroxodiiron(III)-(nta) species shown in Scheme 1 [15-17]; the reactivity of peroxide ion in the (μ-η1:η1)-peroxodiiron(III) species has been

Figure 3. Crystal structure of [Fe2O(nta)2(CO3)]4– ion.
confirmed to be similar to that of singlet oxygen (1Δg) by our studies [18,19], and the negligible activity by the (pac)-compound can be attributed to the inertness of the carbonato chelate, preventing the formation of a (μ- η1:η1)-peroxodiiron(III) species even in the presence of hydrogen peroxide. In the case of the (edda)-compound, the carbonato ion of the dimeric unit readily releases from the compound in the reaction with hydrogen peroxide, to form a dimeric species with a linear Fe-O-Fe core, and finally leading to the formation of a monomeric species [16,20]. For the monomeric compounds, the formation of a (μ-η1:η1)-peroxodiiron(III) species is impossible.
2.2. Structural and Solution Chemical Properties of Fe(III)-(ida) Compound
When the Fe(III)-(ida) was isolated as orange crystal from the solution containing ferric chloride and iminodiacetic acid at pH ~ 7.0 under 25˚C, it consists of a dimeric unit with a linear Fe-O-Fe bridge, as illustrated in
Figure 4 [13] (hereafter we would like to use R = [ida]/ [Fe3+] in the preparation of the iron(III) solution; generally the solutions with R = 4 were employed). Two iminodiacetic acid molecules are coordinated to one iron(III) ion; one acts as a tridentate ligand, and another, as a bidentate ligand (in the discussion on the crystal structure, R’ = [ida]/[Fe3+] was used to characterize the structure; R’ = 2 for the complex illustrated in Figure 4 and it should be noted here that R’ and R are different from each other).
The TBARS formation detected in the solution containing hydrogen peroxide, iron(III) ion and iminodiacetic acid with the ratio of R = 1.2 - 3.2, are shown in Figure 5 [21]. The TBARS formation is negligible when R = 3.2 in the pH range 7 - 8.0, on the other hand, in the solutions of R = 1.2 TBARS formation is notable, and not dependent on the pH (7 - 8). As the TBARS formation does not proceed when the peroxide ion is not coordinated to an iron(III) ion, almost all the iron(III) species in the solution of R = 3.2 with pH 7 - 8 should be a dimeric structure with R’ = 2 illustrated in Figure 4,

Figure 4. Crystal structure of [Fe2O(ida)4]4– ion (R’ = 2).

Figure 5. Absorption spectra of the solution containing iron(III)-(ida) complex, ribose and hydrogen peroxide, treated by TBA (=2-thiobarbituric acid). The absorbance at 532 nm represents the formation of TBARS. blue; R = 1.2, pH 7.0 pink; R = 3.2, pH 7.0 green; R = 1.2, pH 8.0.
where there is no space for hydrogen peroxide to approach to the iron(III) ions. Thus, it seems reasonable to speculate that in the solutions of R = 1.2, the main species is the oxo-bridged dimeric iron(III) species with R’ = 1, Fe2O(ida)2(H2O)4, which is consistent with the mass spectral data. These are demonstrating that there is an equilibrium in the solutions of Fe(III)-(ida) shown in Eq.1.
 (1)
(1)
When the Fe(III)-(ida) solution with R = 4 at pH = 7 ~ 8 was heated to above 40˚C, brown precipitates, maybe Fe(OH)3, occurred. This may demonstrate that the Fe-OFe structure with R’ = 2 species (Figure 4) is less stable than the corresponding R’ = 1 species, and this has been confirmed by the crystal structural data of the related compounds [21].
In the rats administered by the iron(III)-(ida) solution with R = 4, it is reasonable to speculate that concentrations of the iron(III) ion, (ida), and carbonato ion, are diluted with time. As the equilibrium of the Eq.1 is dependent on the concentration of the (ida), the concentration of the species with R’ = 2 may decrease and that of the R’ = 1 may increase with time. In the beginning stage of the administration, since the temperature of the Fe(III)-(ida) solution was raised above 35˚C, and thus we can assume that Fe(OH)3 may form, which is consistent with the report by Mizuno; this may explain the fact that tabular injury by the administration of the Fe(III)-(da) solution with pH 8.0 and R = 4 is negligible [13]. In the cases of the solution of the R = 1.2 with pH 7 - 8, the iron(III) ions in the chelate can bind with the hydrogen peroxide to form a (μ-η1:η1)-peroxodiiron(III) species, because the main species present in the solution should be that with R’ = 1. The validity of the our conclusion on the formation of (μ-η1:η1)-peroxodiiron(III) specie was confirmed by using several iron(III) compounds including (Hedta) [22] and others [13,21].
2.3. Why the Renal Carcinoma Occurs Only in the Vicinity of Renal Proximal Tubules
As stated before, the proximal tubules necrosis and renal carcinoma induced by iron(III)-(nta) and other related compounds are observed only in the renal proximal tubules, but any injury was not observed in the distal position, although much iron(III) ions are present in that position [13]. It should be noted here that the glutathione cycle is highly acting in the renal proximal tubules [23], and this may demonstrate that glutathione cycle is closely related with the formation of (μ-η1:η1)-peroxodiiron(III) species in Scheme I.
At first, we considered the interaction between an iron(III) chelate and the protein. It is generally recognized that transferrin carries an iron (III)ion in the human body,

Scheme 1. Formation of (μ-η1:η1)-peroxodiiron(III) species.
however, the detailed mechanism of up-take of iron ions by apo-transferrin remains unclear at present. In order to clarify the mechanism of iron-ion uptake by apo-transferrin, we have investigated the interactions between several iron(III) chelates and apo-transferrin. The formation of holo-transferrin was checked by ESR spectra, absorption spectra, and capillary electrophoresis [20].
As the results, it has become apparent that the transfer of the iron(III) ion from the iron(III)-chelates to apotransferrin is highly dependent on the structure of the iron(III)-chelates [20]; demonstrating that not all the iron(III) ions in plasma are transferred to apo-transferrin! Strange to say, its trend is very compatible with that in the Table 1, i.e., not all the iron(III) ion are toxic to human body. For example, for the two alkoxo-bridged binuclear iron(III) complexes, Fe2(hida)2(H2O)2 [24] (see Figure 6) and Fe2(HPTP)Cl4 [25] (see Figure 7), the iron(III) ions of only the latter complex are readily transferred to apo-transferrin, but the those of the former complex, are not. Our results have lead to the conclusion that formation of Intermediate shown in Scheme 2, where the two iron(III) ions and the surface of the protein interact via two-point interaction, is necessary for the facile transfer of the iron(III) ions from the iron(III) chelates to apo-transferrin. In the case of Fe2(hida)2(H2O)2, two iron(III) ions cannot interact with the protein via two-point interaction as described in Scheme 2 because of the steric requirement of this complex (Figure 6); on the other hand the two-point interaction is possible for the Fe2(HPTP)Cl4 complex, because the four chloride ions (see Figure 7) are labile in the aqueous solution.
In 1973, Bates et al. reported that iron(III) ions of the Fe(III)-(nta) are readily transferred to apo-transferrin [26], and this may be explained as follows; as mentioned before the carbonato ion of the binuclear complex, Fe2O(nta)2(CO3) is labile, and it may readily dissociate from the complex in the reaction with apo-transferrin, and the two-point interaction between two iron(III) ions and apo-transferrin readily occurs (see Scheme 2).
When green crystals of Fe(III)-(edda) complex was mixed with apo-transferrin in buffer solution, the transfer of the iron(III) ions to apo-transferrin is largely reduced compared with that of Fe(III)-(nta) chelate [20]. This may be elucidated as follows; the color of the buffer is solution containing green solid Fe(III)-(edda) complex

Figure 6. Crystal structure of [Fe2(hida)2(H2O)2].

Figure 7. Crystal structure of [Fe2(HPTP)(Cl)4]+ ion.

Scheme 2. Up-take of iron(III) ion by apo-transferrin.
is light yellow, illustrating that concentration of the green binuclear iron(III) species with bent Fe-O-Fe unit is greatly decreased; this should be due to the dissociation of the dimeric compounds to monomeric species, which was confirmed by the ESR spectrometry. It is clear that monomeric species cannot interact with apo-transferrin through the two-point interaction shown in Scheme 2, and this explains the fact that the transfer of the iron(III) ions to apo-transferrin is largely reduced in the solution of Fe(III)-(edda) chelate. The similar dissociation of a dimeric species to a monomeric one may proceed when the green Fe(III)-(edda) solution was administrated into rat, because the concentration of the ligand (edda) decreases with time.
Next, we considered the interaction between the iron(III) chelates and glutathione cycle. According to the discussions described above, it seems quite likely that some iron (III) chelates which interact with apo-transferrin through the two-point interaction, can interact with glutathione cycle in the same manner. As the glutathione cycle acts as a reductase, it should be remembered here that some binuclear iron(III) compounds exhibit very unique reactivity in the presence of both oxygen and reducing agent, so-called Nishida’s reaction. For example, the binuclear iron(III) complex with H(HPTB), Fe2 (HPTB)(OH)(NO3)2 [21] (see Figure 8) exhibits high activity for oxygenation of linolenic acid in the presence of oxygen, and high catalytic activity for the two-electron transfer reaction to oxygen (formation of hydrogen peroxide) from TMPD (N,N,N’,N’-tetramethyl-p-phenylenediamne) [27,28]. This has been elucidated on the assumption that a binuclear iron(III)-oxygen intermediate formation is promoted through the interaction with linolenic acid or TMPD as shown in Scheme 3 [27-29].
Although the detailed mechanism of the above reactions remains unclear, it is reasonable to assume that the iron(III)-chelate which can interact with apo-transferrin through two-point interaction, can also interact with oxygen in the presence of reducing agent via forming a FeO-O-Fe skeleton (see Scheme 3). In fact, the binuclear iron(III) complex, Fe2(HPTP)(OH)(NO3)2, can oxygenate the linolenic acid in the presence of oxygen, and this complex exhibits high activity for tissue injury when administrated in rat. As it has been exemplified that Fe2(HPTP)(OH)(NO3)2 shows high activity for formation of hydrogen peroxide in the presence of TMPD [28], it

Figure 8. Structure of Fe2(HPTB)(OH)(NO3)2 compound.
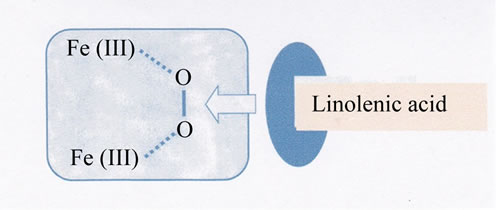
Scheme 3. Formation of Intermediate, Fe2(III)- O2-Linolenic acid.
seems reasonable to speculate that the high tissue damage by Fe2(HPTP)(OH)(NO3)2 should be due to the formation of Fe-O-O-Fe species of the binuclear compound with the reaction of glutathione cycle in the presence of oxygen.
Based on the above discussion, the tissue damage and renal carcinoma by the Fe(III)-(nta) chelate can be explained as follows (see Scheme 4) [20,21]; when binuclear Fe(III)-(nta) compound reacts with glutathione cycle, the carbonato ions release from the compound, leading to the interaction between two iron(III) atoms and the protein. At this stage, when oxygen is present, formation of oxygen adduct, Fe-O-O-Fe may be accelerated through the interaction with the glutathione cycle (see Scheme 3), and the subsequently formed peroxide adduct of the binuclear Fe(III)-(nta) complex shows high oxidative reactivity towards the proteins and DNA [3,20], leading to the tissue injuries and renal carcinoma. In the case of Fe(III)-edda chelate, the main species in the solution is not a dimeric one as described in 2-1, and thus the formation of oxygen adduct, Fe-O-O-Fe in Scheme 3 is greatly reduced, which leads to the lower activity of this complex for oxidative damage. Our conclusion on the active species to induce tissue damage and renal carcinoma (see Scheme 4) can explain comprehensively all the results demonstrated in Table 1 and others not cited in the Table. In the many previous papers, the role of the hydroxyl radical to induce the tissue damage and renal carcinoma has been frequently pointed out [14], but this cannot explain the lower activity in the tissue damage by the Fe(III)-(edda) chelate, and also the difference in the tissue damage observed between the two alkoxo-bridged binuclear iron(III) complexes, Fe2(hida)2(H2O)2 and Fe2(HPTP)Cl4, and also between Fe(III)-(nta) and Fe(III)- (pac) chelates.
In this section, we have demonstrated that the iron toxicity by the artificial iron(III) chelates is mainly due to the formation of (μ-η1:η1)-peroxodiiron(III) species (Scheme 1), and this can be derived from the binuclear iron(III) species, reducing agent (glutathione cycle) and oxygen. But, this toxic species readily generates when hydrogen peroxide is present in the solution containing binuclear iron(III) chelates [18,19]. Thus, we must pay

Scheme 4. Formation of Fe2(III)-O2 species by glutathione reductase.
attention to the presence of hydrogen peroxide in the plasma, which will be discussed in the next section (see Sections 3 and 4). Anyway, it is impossible to overemphasize the importance of the Nishida’s reaction, i.e., some binuclear iron(III) compounds can bind with both the oxygen and hydrogen peroxide, and activate them.
3. NTBI AND H2O2 IN THE PATIENTS OF SEVERAL DISORDERS
3.1. Neurodegenerative Disorders, NTBI and Hydrogen Peroxide
Abnormally high levels of iron (NTBI) in the brain have been demonstrated in a number of neurodegenerative disorders, including Parkinson’s disease and Alzheimer’s disease (AD), and oxidative stress closely related with the increased iron levels in the brain and possible also from defects in antioxidant defense mechanisms is widely believed to be associated with neuronal death in these diseases [3,30,31], but a key question—why do iron levels increase abnormally in some region of the brain?—has not been answered. The recent study have implied that accumulation of large excess of Al and Mn ions induces the abnormal accumulation of NTBI in brain [3,32].
Aceruloplasminemia, originally called familial apoceruloplasmin deficiency, is an iron metabolic disorder where ceruloplasmin deficiency is caused by a lack of apoceruloplasmin biosynthesis and copper metabolism is not disturbed [33]. Ceruloplasmin is expressed in the central nervous system as well as in visceral organs and functions in brain iron metabolism. This disease is characterized by marked iron accumulation in the brain as well as visceral tissue despite low serum iron levels. These findings distinguish clearly aceruplasminemia from hereditary hemochromatosis.
Iron is incorporated into metabolically active compounds or sequestered within ferritin. Ferritin is the principal protein for iron storage and detoxification [4,5]. Almost all cells contain ferritin, which functions both as a safe storage site for iron and as a readily accessible reserve for iron acquired by the cell in excess of its intermediate metabolic needs. The heavy subunit has a ceruloplasmin site which functions in the oxidation of ferrous ion to a ferric form with formation of water, where the ferrous ion released from the ferritin should be chelated compound with amino acids, peptide, or citrate. Thus, the most important problem to be solved is to clarify how the ferrous ion is oxidized to a ferric ion in the patients of aceruloplasminemia. Very recently, we have observed that apo-tarnsferrin exhibits oxidase-like furnction towards several metal chelates [32], suggesting that the ferrous ions are oxidized to a ferric state by apotransferrin with the formation of hydrogen peroxide in the patients of aceruloplasminemia. In the catalytic oxidation of Fe(II) to Fe(III) by ceruloplasmin, hydrogen peroxide does not form. It should be noted here that much quantity of hydrogen peroxide formation occurs in the patients of aceruloplasminemia.
We have reported that hydrogen peroxide catalyzes the formation of di-μ-oxo-diiron(III) species ((B) in Scheme 5) from several iron(III) chelates with amino acid or citrate in the presence of reducing agents such as phenol or alcohol [34], and that this species is derived from (μ-peroxo)(μ-oxo)-diiron(III) species ((A) in Scheme 5) formed in the reaction mixture, which turned to the di-μ-oxo-diiron(III) species through oxidizing the organic molecules such as alcohol or phenols (see Section 2-1). The further aggregation of the di-μ-oxo-diiron(III) species may proceed to give the iron deposition (see also Section 4-2). As these iron(III) ions in the polymeric compounds are not transferred to apo-transferrin [20], above discussion explains the marked iron accumulation in the brain as well as visceral tissue despite low serum iron levels observed [33], and the fact that highly toxic (μ-peroxo)(μ-oxo)-diiron(III) species ((A) in Scheme 5) generated under the presence of hydrogen peroxide, can degrade the peripheral proteins or DNA, etc. [3] is highly consistent with the oxidative damages observed for the patients of aceruloplasminemia [33].
3.2. Disorders Due to Abnormal Ferritin
Iron is essential to all the livings, but to prevent its toxicity it must be associated to specialized molecules. Of those, ferritins play special roles because of their ubiquitous distributions in all tissues, to the tight iron-dependent gene expression, and to their capacity to storage amount of iron (up to 4000 Fe atoms per molecule) inside the large protein shell, in a nontoxic and bioavailable form [4,5]. Mammalian ferritins are heteropolymers made of two different subunit types named Hand L-chain with equivalent proportion. The H-chain carries a ferroxoidase center which appears to be essential for iron corporation, whereas the L-chain facilitates iron mineralization inside the cavity (see Eq.2) [4,5].

Scheme 5. Formation of Fe(III) deposition from iron(III) chelate by H2O2 and reducing agent.
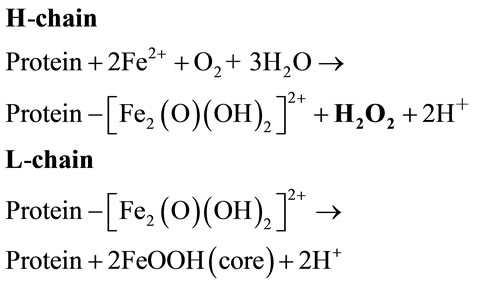 (2)
(2)
It is well known that in the H-chain, two iron(II) ions are oxidized to an iron(III) state through the formation of (μ-η1:η1)-peroxodiiron(III) species in Scheme 2; this process is almost the same as observed for the reactions in RNR (ribonucleotide reductase) [35] and MMO (methane monooxygenaze) [36]. Although the hydrogen peroxide generated in this step is used for the oxidation reactions of the organic compounds in RNR and MMO, the hydrogen peroxide formed in the H-chain is only released from the site! This difference of the reactivity of peroxide ion in the (μ-η1:η1)-peroxodiiron(III) species among the enzymes has been elucidated by us [9,10, 34,37]. Anyway, it is apparent that hydrogen peroxide formed in H-chain is not used, only released from the site.
We have pointed out that the hydrogen peroxide released from H-chain is necessary in the L-chain for the mineralization of the iron cores through the forming a Fe-oxo bonding as illustrated in Scheme 5 [34], although the role of hydrogen peroxide in the L-chain has been completely neglected in the previous papers [4,5]. We also demonstrated that rich carboxyl groups exposed on the active surface of L chain is necessary for the normal iron nucleation and mineralization on the basis of results on the crystal structural determination of several iron(III) compounds [34], and have pointed out that the precise formation of heteropolymers by the Hand L-chains with equivalent proportion is essential for the utilizing of the peroxide ion; this is the main reason for the fact that ferritin is non-toxic.
Although Hand L-chains are encoded by different genes, synthesis of both chains is controlled by a common cytosolic protein, iron regulatory protein (IRP), which binds to the iron responsive-element (IRE) in the 5’-UTR of the Hand L-chain mRNAs. Kato et al. showed that a single mutation in the IRE region of H-chain mRNA induces the decrease of H-chain, and affected dominantly inherited iron overload, leading to tissue iron deposition, and also to impairment of the ferroxidase activity by H-chain [38]. Hereditary hyperferritinemia-cataract syndrome (HHCS) is an autosomal and dominant disease caused by heterogeneous mutations in the IRE of the 5’-untraslated flanking region of ferritin L-chain mRNA. According to the results by Levi et al., L-chain levels are up to 20-fold higher in HHCS than in control cells, and were not affected by iron supplementation or chelation [39]. Above facts are implying that mutations in mRNAs induce the unbalance between Hand L-chains, leading to the abnormal presence of hydrogen peroxide in the ferritin, which leads to the formation of iron deposition as illustrated in Scheme 5, and also to severe oxidative damage as observed for the patients reported by Kato et al. and Levi et al.
Neuroferritinopathy typically presents with progressive adult-onset chorea or dystonia affecting one or two limbs, and subtle cognitive deficits [40]. The movement disorder involves additional limbs within five to ten years and becomes more generalized within 20 years. The majority of individuals develop a characteristic orofacial action-specific dystonia related to speech that leads to dysarthrophonia. Cognitive deficits and behavior issues become major problems with time. Neuroferritinopathy is a neurodegenerative disease caused by the mutations in the ferritin L-chain gene [40], and thus it seems quite reasonable to assume that the presence of excess iron accumulation including iron deposition and oxidative damage observed for the patients should be due to the break of equivalency between the Hand L-chains in the ferritin, as pointed out for hereditary hyperferritinemia-cataract syndrome (HHCS).
4. ELIMINATION OF NTBI BY THE USE OF SUPER-POLYPHENOLS AND ZINC(II) ION
4.1. Polyphenols and Super-Polyphenols as Antioxidants
It is well known that catechins which are contained in the Japanese green tea react with a Fe(III) ions to form deeply-colored (violet) species, and we also observed that the iron(III) chelates with (nta), (ida) and others give a deeply-colored species in the reaction with catechins In the tris-catecholato-iron(III) species, there is no space where hydrogen peroxide or oxygen molecule can coordinates to iron(III) ion, thus these catechins may depress the oxidative damage due to the NTBI in addition to their antioxidative activity to destroy the radicals, which is consistent with that Japanese tea catechins exhibit high preventing effects against the lifestyle-related diseases [41].
Based on these facts, we have synthesized new super-polyphenols, which contain more than 100 molecules of catechol derivative in one polymeric compound (see Scheme 6) [32]; these are sometimes water-soluble, but in another cases, water-insoluble. We found that some of these compounds can eliminate NTBI effectively from the plasma, and thus we can hope that these compounds should depress the oxidative stress due to NTBI in human body as one of the most hopeful substances for labile iron removal therapy (LIRT) [42]. At present, we

Scheme 6. Illustration of super-polyphenols.
have plan to delete both the non-toxic and toxic watersoluble NTBI at the same time by our super-polyphenols of water-insoluble form, and this project is now under progress in Japan.
In addition to the elimination of NTBI, we must delete the hydrogen peroxide in the plasma (see Section 3). Very recently we have observed that some derivatives of our super-polyphenols exhibits high ability for the removal of H2O2 from the solution, and thus I hope that our super-polyphenols in Scheme 6 will be used over the whole world in future for the prevention and treatment of many diseases related with NTBI.
4.2. Role of Zinc(II) Ions for Formation of Iron Deposition and Its Antioxidative Function
We have found that iron deposition readily occurs on the aggregates of amyloid beta-peptide (Aβ (1-40) when zinc(II) chloride solution is added to the solution (pH = 7.4) containing iron(III) compounds with (nta), (edda), and other amino acid derivatives, and Aβ (1-40) [43]. These facts are implying that iron(III) compounds with amino acids or peptides in plasma may be an intrinsic iron(III)-ion carrier to induce the high level accumulation of iron(III) ions in the amyloid deposits.
The formation of iron deposition reported by us may proceed according to the Scheme 7: zinc(II) complex which contains hydroxide ion (OH–) [44] approaches to the protein, where iron(III) complex binds with the protein through two-point interaction as illustrated in Scheme 2, and then the transfer of the hydroxide ion from the zinc(II) to the iron(III) ion occurs, leading to the formation of di-μ-oxo-diiron(III) species (see Scheme 5) and finally to the iron deposition as demonstrated in Section 3-1. Above discussion seems to be consistent with the facts that the total zinc(II) concentration is relatively reduced compared with that of normal cases, and massive iron deposition are observed in the brain and on several organs such as kidney or spleen of the aceruplasminemia patients [33], amyloid deposits contains generally much quantities of iron(III) and zinc(II) ions [31], and that alternations in zinc homeostasis have been reported in Parkinson’s and Alzheimer’s diseases as well as in transient forebrain ischemia, seizures and traumatic brain injury, where high level of NTBI have been re-

Scheme 7. Binuclear iron(III) and Zn(II) species on the surface of protein.
ported [45].
Alzheimer’s disease is a progressive neurodegenerative disorder clinically characterized by cognitive dysfunction. The major pathological hallmark of AD is deposition of amyloid β-protein (Aβ) in the brain [31]. But, it has been recently suggested that the toxicity of Aβ and other amyloidogenic proteins lies not in the insoluble fibrils that aggregate but rather in the soluble oligomeric intermediates, indicating that the soluble oligomers may be more important pathologically than are the fibrillar deposits [46,47]. Based on the facts that there is considerable evidence that the Aβ-deposition in Alzheimer’s disease is induced by zinc(II) ion [45] and amyloid deposition generally contain much iron(III) and zinc(II) ions [31], and since we are considering that origin of the high toxicity by soluble Aβ oligomers should be due to the iron(III) species bound in the oligomers on the basis of the discussions described in Section 2, it seems reasonable to consider that the amyloid deposition formation, which deletes the toxic NTBI from the plasma, should be one of the antioxidative function by zinc(II) ion [43]. If our proposition that origin of the high toxicity by soluble Aβ oligomers should be due to the iron(III) species bound in the oligomers is right, it seems likely that our new water-soluble super-polyphenols can protect the oxidative stress due to NTBI in oligomeric Aβ.
Very recently, the possibility of altering zinc(II) levels within the brain is emerging as a new target for the prevention and treatment of psychiatric and neurological diseases [48]. But it should be strongly noted that zinc(II) ion can cleave DNA hydrolytically under the presence of hydrogen peroxide [44], and thus excess zinc(II) ions are very toxic in the brain.
5. SUMMARY
We have been interested in the elucidation of oxygen activation mechanism in biological oxygenases [49], and have reached to the conclusion that 1) in the case of mono-oxygenases, O2 is changes to the peroxide ion, and the activation of this peroxide ion proceeds through the coordination to the iron atom [9,10]. On the other hand, O2 directly interacts with the iron ion in the di-oxygenases, and it is activated without the change of the oxidation state of the metal ion [9,10], 2) the electronic features of the substrate and peripheral organic groups (including the co-enzymes) highly contribute to determine the reactivity of the activated oxygen derivative, and 3) in both oxygenases, the activated oxygen derivative acts an electrophile, its reactivity being very similar to that of singlet oxygen (1Δg) [3,9,10]. According to the results described in this review, it is quite clear that the intrinsic factors to induce oxidative stress should be the iron ions in the labile iron pool, i.e., some of the watersoluble NTBI react with oxygen or hydrogen peroxide, changing these molecules to those exhibiting the reactivity similar to singlet oxygen (1Δg), and this is the main reason for NTBI to induce the oxidative stress [3,50].