Use of Perovskite-Type Lanthanum Nickelate Synthesized by the Polymeric Precursor Method in the Steam Reforming Reaction of Methane ()
1. Introduction
Worldwide, environmental problems, such as air pollution resulting from the emission of gases into the atmosphere, are being discussed and debated through conferences and meetings of rulers several countries in order to create a strict environmental policy to control and limit the quantities of gases emitted. Gases such as NO, CO, CxHy, and SOx, and contribute to the greenhouse effect, have power to destroy the ozone layer. One of the great landmarks in an attempt to minimize the amount of gases released into the atmosphere was the mandatory use of catalyst (containing noble metals such as palladium, platinum and ruthenium) in automotive vehicles, converting the gases of incomplete combustion to CO2, H2O and N2.
In recent years, the Perovskite type oxides (general formula ABO3) have been recognized as active catalysts, instead of the noble metals, for a variety of reactions, especially in environmental catalysis, such as exhaust gas treatment, catalytic combustion of hydrocarbons, oxidation of CO and hydrocarbons, NO reduction with CO, reactions and retirement [1-4].
The steam reforming reaction has been employed in industrial catalytic processes to produce H2 and syngas [5]. NiO/Al2O3, which is a well-known industrial catalyst, is effective for steam reforming of methane or natural gas fuels [6,7]. However, this catalyst frequently forms deposits of carbon, which are harmful to the process, once they deactivate the metallic catalyst, thus decreasing its service time [8]. Ionic oxides with perovskite structure, supported or not, are potential oxidation catalysts and promising candidates for the steam reforming reaction, because the Ni present in the perovskite structure can be reduced to its metallic state and highly dispersed in a solid oxide.
A perovskite-type mixed oxides have the ABO3 crystal structure where cations with large ionic radii are coordinated to 12 oxygen atoms and occupy the A sites. Cations with smaller radii are coordinated to 6 oxygen atoms and occupy the B sites. The A sites correspond to the center of the cube whereas the B sites to octahedral positions [9]. The perovskite lattice can accommodate multiple cationic substitutions with slight structural changes. The properties of this structure are mainly determined by the B sites which can be partially substituted [10]. These solids show interesting optical, magnetic, catalytic, dielectric and conduction properties, of potential importance for the application of these materials in new technologies [11,12]. The formation of perovskite-type oxides only occurs when the ratio between the radii of the metal ions involved complies with the tolerance factor t, which should be between 0.75 to 1.0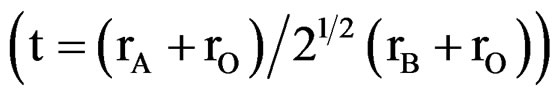 . One of the major properties of these oxides is the possibility of partial substitution of cations A and/or B, leading to a large class of materials of general formula
. One of the major properties of these oxides is the possibility of partial substitution of cations A and/or B, leading to a large class of materials of general formula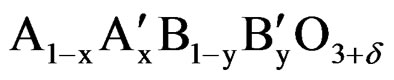 . In the formula, δ represents the excess or deficiency of oxygen due to nonstoichiometry these species. The partial substitution of A and/or B for metals with different oxidation states generates defects in the structure (anionic or cationic vacancies), which are usually associated with physical and chemical properties of the material, favoring, for example, ion transport within the structure, leading to interesting differences in catalytic performance.
. In the formula, δ represents the excess or deficiency of oxygen due to nonstoichiometry these species. The partial substitution of A and/or B for metals with different oxidation states generates defects in the structure (anionic or cationic vacancies), which are usually associated with physical and chemical properties of the material, favoring, for example, ion transport within the structure, leading to interesting differences in catalytic performance.
In the present work, we prepare perovskite type oxides by the polymeric precursor method [12]. This method presents several advantages compared with other approaches, such as high product homogeneity and purity, uniform distribution of phases and relatively low processing temperatures [13,14]. Both supported and unsupported catalysts were characterized by X-ray diffraction, BET surface area and thermo-programmed reduction. The activity of the catalysts in the methane steam reform was also evaluated.
2. Experimental
2.1. Synthesis of the LaNiO3 by the Polymeric Precursor Method
LaNiO3 was synthesized by the polymeric precursor method. A nickel citrate solution was prepared from Ni(NO3)3·6H2O and citric acid in a molar ratio of 1:1.5 under constant stirring at 60˚C for 30 min. A stoichiometric amount of La(NO3)2·6H2O was mixed with the nickel citrate solution at 70˚C for 30 min. The temperature was slowly increased to 90˚C and ethylene glycol was added in the ratio 60:40 (citric acid to ethylene glycol). The solution was stirred at that temperature for 1 hour. The gel formed was calcined at 300˚C for 2 hours resulting in the precursor powder denominated LN3. This material was calcined at 800˚C for 4 hours and denominated LN8 (LaNiO3-800˚C). The precursor powder was supported on alumina or zirconia by the impregnation method and calcined at 900˚C for 4 hours. The mass proportion of perovskite:support used was 1:3. Therefore, the resulting material denominated LN3A9 was treated at 300˚C, supported on alumina and calcined at 900˚C for 4 hours. On the other hand, LN3Z9 stands for the material treated at 300˚C, supported on zirconia and calcined at 900˚C for 4 hours.
2.2. Characterization of the LaNiO3
Thermogravimetric analyses were carried out in a Shimadzu TG/DTA-60H instrument at a heating rate of 10˚C·min–1 in air flowing at 50 cm3·min–1. X-ray diffraction patterns were obtained from a Shimadzu XRD-6000 diffractometer, used to scan the angular range 10˚ £ 2q £ 80˚ with CuKa radiation (l = 1.5418 Å). The surface area of the powders was measured by nitrogen adsorption on a NOVA 2000 system. The catalysts were characterized by temperature programmed reduction (TPR) profiles recorded by heating about 20 mg of the samples from 50˚C to 1000˚C in an AutoChem II 2920 instrument at a heating rate of 10˚C·min–1 under H2-Ar (10% H2) on a quartz reactor. Catalytic tests were accomplished in a fixed bed continuous flow microcatalytic reactor. The conditions used were: inlet temperature 600˚C; reaction temperature 750˚C; pressure of 6 bar; dynamic flow of 150 mL·min–1 (for CH4), 0.27 mL·min–1 (for H2O); catalyst mass = 5.73 g; H2O/CH4 molar ratio of 2.5 and reaction time of 4 hours.
3. Results and Discussion
X-ray diffraction analysis revealed that the precursor powder was amorphous. The formation of a crystalline phase can be seen from the X-ray diffractogram of the perovskite catalyst calcined at 800˚C for 4 hours (LN8, Figure 1(a)) and the perovskites supported on alumina or zirconia (LN3A9, Figure 1(b) and LN3Z9, Figure 1(c)).
The LN8 catalyst is a single-phase material with perovskite structure characterized by intense reflections in 2q equals to 33˚, 48˚ and 59˚. All reflections were indexed as a rhombohedral cell, R3c space group (JCPDICDD 34-1028). The X-ray diffractograms of the LN3A9

Figure 1. XRD patterns (a) LN8, and (b) the perovskites supported on alumina or zirconia and (c) LN3Z9.
and LN3Z9 catalysts (Figures 1(b) and (c)) suggest that, after the impregnation of LN3 on alumina or zirconia and subsequent calcination, the perovskite structure crystallized on the support. All the peaks shown in the diffractogram correspond to the perovskite structure as well as of the supports (alumina or zirconia). The perovskite structure was confirmed in these materials by its princepal peaks at 2q = 32.74˚ - 33.1˚; 47.2˚ - 47.8˚ and 59.7˚ - 59.9˚.
The thermogravimetric plots of LN3, LN3A9, LN8 and LN3Z9 are shown in Figure 2. The profile corresponding to LN3 (Figure 2(a)) revealed mass losses corresponding to dehydration (30˚C to 180˚C) followed by the decomposition of nitrates (180˚C to 300°C), most of the ionic citrates (COO–) and ethylene glycol (300˚C to 600°C). The remainder ionic citrates are converted into CO2 and CO between 600°C and 800˚C to finally form oxides at higher temperatures. A slight mass gain was observed between 800 and 900˚C due to the oxidation of Ni2+ to Ni3+, likely resulting in lattice vacancies.
The TG profile shown in Figure 2(d) corresponds to the LN3Z9 catalyst. The oxidation of Ni2+ to Ni3+ was also noticed, meaning that in air, the zirconia support did not interfere with the nickel catalytic site. Plain LN8 (Figure 2(c)) depicted higher mass gain corresponding to oxidation compared to catalysts supported on zirconia. Increased nickel oxidation was indeed expected for the unsupported catalyst.
Finally, the TG plot of the alumina supported catalyst (Figure 2(b)) revealed a moss loss of ~5% attributed to the γ → α alumina phase transformation rather than to any effect of the catalyst on the support. The effect of the alumina phase transformation exceeds that of the oxidation of Ni2+, therefore the behavior observed differs from that of zirconia supported catalysts. Specific surfaces areas (SSA) values for LN8, LN3A9 and LN3Z9 were

Figure 2. The Tg curves for (a) LN3, and (b) the alumina supported catalyst, and (c) LN8 and (d) the LN3Z9 catalyst.
31.5, 80.0 and 2.1 m2·g–1. LN8 has SSA values characteristic of perovskite type oxides. The materials supported on alumina (LN3A9) presented higher area values because alumina has a porous structure capable of withstanding a large dispersion of active species.
The Figures 3(a)-(c) show the temperature-programmed reduction (RTP) profile of the LN8, LN3Z9 and LN3A9 samples. The RTP curve shows two reduction peaks, one at around 355˚C and the other at 467˚C. These peaks correspond to successive changes in the perovskite structure. The first peak can be attributed to the formation of the La4Ni3O10 structure according to the following reaction:
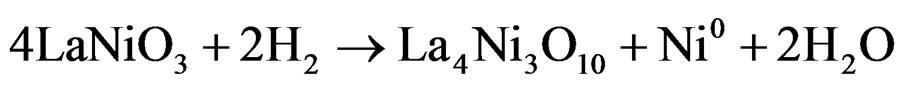 (1)
(1)
The second stage corresponds to the formation of the spinel La2NiO4 phase. Immediately afterwards this phase is reduced to metallic nickel between 400˚C and 530˚C, according to:
 (2)
(2)

According to these RTP profiles, the complete reduction of LaNiO3 into La2O3 and metallic nickel occurs at around 520˚C. In the case of the LN3A9 and LN3Z9 samples, it was observed that they also had two reduction peaks for the changes in the perovskite structure previously discussed, but these peaks were displaced to higher temperatures. Furthermore, it can also be seen that a slight widening occurred in the first reduction peak. It was observed that no significant changes in reduction profile

Figure 3. The RTP for (a) LN8, and (b) LN3Z9 and (c) LN3A.
occurred (intensity and characteristics of the reduction peaks), suggesting that no secondary phases were formed between the perovskite and the support during synthesis.
The results of methane conversion of the LN8, LN3A9 and LN3Z9 catalysts are shown in Figure 4. The calculation of methane conversion was performed using carbon mass balance, excluding the amount retained in coke formation. Therefore, it is considered that all the carbon entering the system in the form of methane must exit as non-converted carbon dioxide, carbon monoxide and methane. The conversion equation can be expressed as follows:
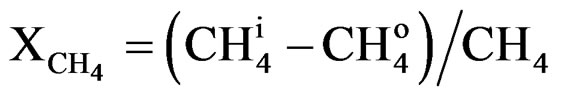 (3)
(3)
where:
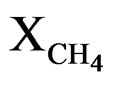 = Methane conversion;
= Methane conversion;
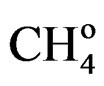 = Amount of methane at the reactor outlet;
= Amount of methane at the reactor outlet;
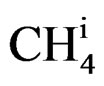 = Amount of methane at the reactor inlet;
= Amount of methane at the reactor inlet;
For carbon mass balance, Equation 4 was used:
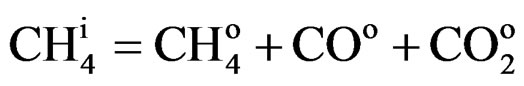 (4)
(4)
where:
COo = Amount of carbon monoxide at the reactor outlet
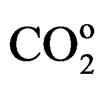 = Amount of carbon dioxide at the reactor outlet According to the results obtained, the alumina supported catalysts (LN3A9) displayed the highest activity, which may be attributed to the additional role of aluminum and by the presence of superficial acid sites in promoting the methane conversion reaction. The presence of a support (alumina or zirconia) is extremely important to maintain catalyst stability during deactivation reaction, and consequently improve methane conversion levels. It was observed that when the catalyst is not supported (LN8), a rapid deactivation occurs at the beginning of the
= Amount of carbon dioxide at the reactor outlet According to the results obtained, the alumina supported catalysts (LN3A9) displayed the highest activity, which may be attributed to the additional role of aluminum and by the presence of superficial acid sites in promoting the methane conversion reaction. The presence of a support (alumina or zirconia) is extremely important to maintain catalyst stability during deactivation reaction, and consequently improve methane conversion levels. It was observed that when the catalyst is not supported (LN8), a rapid deactivation occurs at the beginning of the

Figure 4. The curves of methane conversion for (a) LN8, and (b) LN3Z9 and LN3A9.
reaction, likely due to the deposition of carbon on the catalytic surface, according to the following reaction:
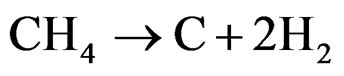 (5)
(5)
This behavior suggests that improved activation of water molecules occurs for LN3A9 and LN3Z9, which favors the removal of carbon deposited at the active nickel sites according to the following reaction:
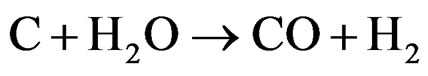 (6)
(6)
The higher activity of LN3A9 compared to that of LN8 and LN3Z9 may also be correlated to its larger surface area and to better nickel dispersion, as can be observed in the results discussed earlier.
The lower methane conversion in the LN3Z9 catalyst compared to LN3A9 may be attributed to lower carbon removal efficiency. One of the differences found in these two catalysts is the amount of oxygen vacancies available on the support surface. The greater availability of oxygen vacancies increases the water molecule activation and allows better carbon removal, keeping the nickel species active for methane decomposition. Methane conversion was 94% for LN3A9, 68% for LN3Z9 and 52.4% for LN8.
Figure 5 shows the selectivity products for the reactions studied using LN8, LN3A9 and LN3Z9. The values found for H2 selectivity were around 74.4%, 70.1% and 64.5% for LN3A9, LN3Z9 and LN8, respectively. With respect to CO and CO2 contents, the LN3A9 and LN3Z9 catalysts yielded similar results, but alumina supported samples showed higher CO formation. The selectivity to reaction products of LN3A9 and LN3Z9 catalysts were quite similar, although the LN3A9 catalyst was more stable during the reaction and also showed higher conversion and selectivity values.
Figure 6 shows the H2, and CO and CO2 output profile after 3.5 h of reaction using LN8, LN3A9 and LN3Z9 catalysts. The amounts of selective products obtained