Green and High Efficient Synthesis of 2-Aryl Benzimidazoles: Reaction of Arylidene Malononitrile and 1,2-Phenylenediamine Derivatives in Water or Solvent-Free Conditions ()
Received 20 August 2015; accepted 6 December 2015; published 9 December 2015


1. Introduction
In recent years, significant attentions have been considered to the organic reaction under aqueous media, particularly from the viewpoint of green chemistry [1] - [4] . Using water, in contrast to common hazardous organic solvents, offers many advantages such as: simplicity of reaction conditions, ease of work-up and product isolation, increasing the selectivity of a wide variety of organic reactions and accelerating reaction rates [5] [6] . Benzimidazole derivatives are important scaffolds in medicinal chemistry due to their biological and pharmacological activities. These compounds exhibit activity against several viruses include HIV [7] [8] , influenza [9] , herpes (HSV-1) [10] , RNA [11] and human cytomegalovirus (HCMV) [12] . They have also been employed as antihypertensive, antiviral, anticancer, antiulcer, antifungal and untihistamine [13] - [18] .
In view of the biological importance of benzimidazoles, there have been growing interests in the development of efficient, fast, simple and environment friendly synthetic methods for the preparation of these molecules. Several procedures have been reported for the synthesis of 2-substituted benzimidazoles: Condensation of 1,2- phenylenediamines with carboxylic acids, acid chlorides, nitriles, imidates and orthoesters under strong acidic conditions, sometimes combined with very high temperatures or useing microwave irradiation, [19] - [22] oxidative cyclodehydrogenation of 1,2-phenylenediamine and aldehydes in the presence of different oxidants [23] - [26] , transition-metal-catalyzed intramolecular cyclization of 2-haloanilides and their analogues [27] - [29] and also the condensation reactions of 1,2-phenylenediamine with β-ketonitriles [30] , β-ketoesters [31] [32] , or β- diketones [33] under microwave radiation and high temperature conditions or in the presence of a catalyst. Although, all of these methods are widely employed, but they have drawbacks such as low yields, the use of expensive and toxic reagents, catalysts and solvents, long reaction times, formation of side-products, tedious work-up procedure, and in some cases, harsh reaction conditions are required. Therefore, development of efficient, economical, and environmentally benign synthetic protocols for their construction is an important goal in diverse areas of chemistry. In addition, a number of other useful green reactions for the synthesis of benzimidazole derivatives have been reported in the literature. For example, Su and co-workers reported synthesis of substituted benzimidazoles from 1,2-phenylenediamine and arylaldehydes or arylmethylenemalononitriles absorbed on silica gel by intermittent grinding or by a microwave-assisted technique under solvent- and catalyst-free conditions [34] . Also, Chikashita and co-workers described formation of 2-aryl benzmidazoles with reaction between of arylidenemalononitriles or β-nitrostyrenes with 1,2-phenylenediamine in ethanol at boiling temperature through a simple and efficient transfer-hydrogenation process from the in situ generated benzimidazolines to activate olfines [35] .
In order to further development of synthetic route of benzimidazoles under green reaction conditions, here, we devoted our effort for the synthesis of 2-aryl benzimidazole derivatives in water as a green solvent as well as solvent-free conditions (Scheme 1).
2. Result and Discussion
In this work, we report a highly efficient, and environmentally benign procedure for the reaction of 1,2-pheny- lenediamine derivatives 1 with arylidenemalononitrile 2 in aqueous medium as a green solvent to produce benzimidazole derivatives 3. Also, in continuation of our goal towards performing of this reaction under another green condition, we have developed reaction between reactants under solvent-free condition using thermal heating method after grinding. Arylidenemalononitrile 2 was reacted with 1,2-phenylenediamine derivatives in the presence of water to produce the related products 3 with excellent yields. We initially employed 1,2-phenyl- nediamine 1a (1 mmol) and arylidenemalononitrile 2a (2 mmol) in water at room temperature as a model reaction. In this condition the reaction wasn’t complete after 14 hours (Table 1, entry 1). Therefore, various conditions have been designed to determine the optimized conditions. Different solvents such as water, ethanol, methanol, acetone, dimethylsuloxide, tetrahydrofuran, and chloroform were explored. Also, the reaction was performed under different temperatures such as 25˚C, 50˚C, 75˚C and 90˚C. The results are summarized in Table 1. As can be seen, the best result was obtained by the reaction mixture in water at 75˚C for 20 min to yield product
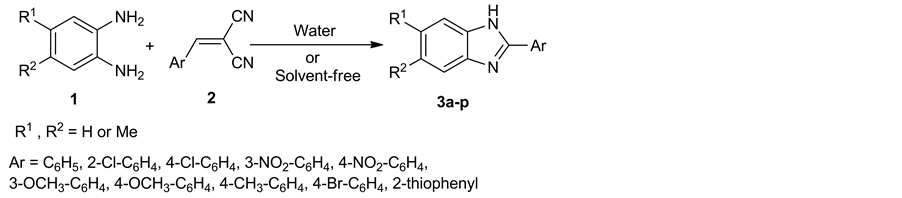
Scheme 1. Synthesis of 2-aryl benzimidazole derivatives.
![]()
Table 1. Effect of different reaction conditions for synthesis of product 3aa.
aReaction condition: 1,2-phenylenediamine 1a (1 mmol) and arylidenemalononitrile 2a (2 mmol); bIsolated yield. cyield based on TLC analysis.
3a (Table 1, entry 9). Although, the reaction gave high to excellent yields in organic solvents, but using water is the most advantageous to this method (Table 1, entries 14 - 19). After optimizing the reaction condition, to explore the scope and generality, the synthesis of benzimidazole derivatives 3a-p were carried out through the reaction of 1,2-phenylenediamine derivatives and a wide diversity of arylidenemalononitrile in high yields (Table 2). Interestingly, we observed that the position and nature of substitution on the ring of arylidenemalononitrile did not make much difference in reactivity, indicating the wide scope of this methodology.
In continuation of this study, we are interested in solvent-free conditions as another green procedure by using grinding method. Thus, we have synthesized a series of 2-substituted benzimidazoles 3a-p by the reaction of reactants under this method on heating. Therefore, the reaction of arylidenemalononitrile 2a and 1,2-phenylne- diamine 1a proceeded successfully in an open vial through grinding of two components together and then heating at 90˚C for 30 min. This reaction started immediately after heating, with liquification of the mixture, followed by solidification of the mixture of reaction. By comparing the reaction time and yields of entries 20 to 22 in Table 1, it was found that 30 min and 90˚C was best conditions for this reaction. Also, it was found that both
![]()
Table 2. Synthesis of 2-aryl benzimidazoles 3a-p.
aReaction condition: 1,2-phenylenediamine derivatives 1 (1 mmol), arylidene malononitrile 2 (2 mmol), water (5 ml) under 75˚C and 20 min; bReaction condition: 1,2-phenylenediamine derivatives 1 (1 mmol), arylidene malononitrile 2 (2 mmol), grinding heating at 90˚C for 30 min; cIsolated yield.
electron-donating and electron-deficient groups were suitable for this reaction because the products were obtained in excellent yields. In addition, a heterocyclic arylidenemalononitrile such as 2-(thiophen-2-ylmethylene) malononitrile could react with 1,2-phenylenediamine and 4,5-dimethyl-1,2-phenylenediamine to afford the corresponding benzimidazole (Table 2). The known compounds were identified by comparison of their melting point with those reported earlier (see references in Table 2). Also, a number of these compounds was characterized by its 1H-NMR. A plausible mechanism based on reported previous work [35] is proposed in Scheme 2. Initially, Michael addition reaction of 1,2-phenylenediamine 1 with the arilydenemalononitrile 2 gave intermediate 4. The consequent proton transfer results transformation of 4 into 5. Then this intermediate converted to benzimidazoline 7 with leave malononitrile as leaving group perhaps by one of two paths (path a or b). The benzimidazole 3 is formed through a simple and efficient transfer-hydrogenation process from in situ generated benzimidazoline to arylidenemalononitrile.
3. Conclusion
In summary, we have reported green and highly efficient method for the synthesis of 2-aryl benzimidazoles in water as well as under solvent-free and catalyst-free conditions. The main advantages of these procedures are environmentally friendly, the operational simplicity, short reaction times, simple work-up procedures and high yields.

Scheme 2. Plausible mechanism for the formation of products 3a-p.
4. Experimental
1,2-phenylenediamine derivatives, malononitrile and aldehyde derivatives were purchased from the Merck and Fulka companies and were used without further purification. Melting points were determined on Electrothermal 9100 apparatus. 1H NMR spectra was recorded on a Bruker Avance 300 MHz employing tetramethylsilane as an internal standard.
4.1. General Procedure for the Synthesis of 2-Aryl Benzimidazole 3a-p in Water
1,2-phenylenediamine (1 mmol) was dissolved in 5 ml water at 75˚C. Then, arylidenemalononitrile (2 mmol) was added to this solution, and immediately the reaction mixture liquefied and resolidified. The reaction was monitored by TLC (petroleum ether: Ethyl Acetate (8:2)) till the disappearance of the starting arylidene malononitrile. After cooling the resultant reaction mixture, recrystalization in ethanol-water and finally pure 2-aryl bezimidazole was filtered out.
4.2. General Procedure for the Synthesis of 2-Aryl Benzimidazole 3a-p in Solvent-Free Conditions Using Conventional Heating Method
Arylidenemalononitrile (2 mmol) and 1,2-phenylenediamine (1 mmol) were mixed thoroughly with glass stirrer and heated at 90˚C. The reaction mixture liquefied and resolidified in 30 min. Completion of the reaction was checked by TLC (petroleum ether: Ethyl Acetate (8:2)). After cooling the resultant semi-solid reaction mixture, crystallization was performed in ethanol-water and 2-aryl bezimidazole was filtered out.
2-Phenylbenzimidazole [36] : m.p. = 287˚C - 289˚C, 1H NMR (300 MHz, DMSO-d6): δ = 7.19 - 7.25 (m, 2H, ArH), 7.48 - 7.54 (m, 1H, ArH), 7.62 (d, J = 8.6 Hz, 2H, ArH), 7.67 - 7.73 (m, 2H, ArH), 8.17 (d, J = 8.6 Hz, 2H, ArH), 8.54 (s, 1H, NH).
2-(2-Cholorophenyl) benzimidazole [41] : m.p. = 231˚C - 233˚C. 1H NMR (300 MHz, DMSO-d6): δ = 7.26 - 7.36 (m, 2H, ArH), 7.38 - 7.44 (m, 2H, ArH), 7.48 - 7.51 (m, 1H, ArH), 7.69 (m, 2H, ArH), 8.41 (m, 1H, ArH), 10.36 (br s, 1H, NH).
2-(4-Chlorophenyl) benzimidazole [37] : m.p. = 291˚C - 93˚C, 1H NMR (300 MHz, CDCl3): δ = 7.29 - 7.32 (m, 1H, ArH), 7.49 - 7.54 (m, 3H, ArH), 7.73 - 7.88 (m, 3H, ArH), 7.98 - 8.00 (m, 1H, ArH), 9.3 (br s, 1H, NH).
2-(3-Nitrophenyl) benzimidazole [38] : m.p. = 204˚C - 207˚C, 1H NMR (300 MHz, DMSO-d6): δ = 7.20 - 7.30 (m, 2H, ArH), 7.57 (d, J = 7.3 Hz, 1H, ArH), 7.71 (d, J = 7.8 Hz, 1H, ArH), 7.85 (dd, J1 = 7.9 Hz, J2 = 7.9 Hz, 1H, ArH), 8.32 (dd, J1 = 2.0 Hz, J2 = 7.9 Hz, 1H, ArH), 8.61 (d, J = 7.9 Hz, 1H, ArH), 9.01 (dd, J = 2.0 Hz, J = 2.0 Hz, 1H, ArH), 13.30 (s, 1H, NH ).
2-(3-Methoxyphenyl) benzimidazole [41] : m.p. = 202˚C - 205˚C, 1H NMR (300 MHz, DMSO-d6): δ =3.84 (s, 3H, OMe), 6.95 (d, J = 8.6 Hz, 2H, ArH), 7.00 - 7.92 (m, 4H, ArH), 8.03 (d, J = 8.6 Hz, 2H, ArH).
2-(4-Methoxyphenyl) benzimidazole [38] : m.p. = 223˚C - 226˚C, 1H NMR (300 MHz, DMSO-d6): δ = 3.83 (s, 3H, OMe), 6.95 (d, J = 8.8 Hz, 2H, ArH), 7.23 (dd, J1 = 3.2 Hz, J2 = 6.0 Hz, 2H, ArH), 7.60 (dd, J1 = 3.2 Hz, J2 = 6.0 Hz, 2H, ArH), 8.04 (d, J = 8.8 Hz, 2H, ArH).
2-(Thiophen-2-yl) benzoimidazole [40] : m.p. = 330˚C - 333˚C, 1H NMR (300 MHz, CDCl3): δ = 7.15 - 7.18 (m, 1H, ArH), 7.27 - 7.29 (m, 2H, ArH), 7.47 - 7.49 (m, 2H, ArH), 7.61 - 7.62 (m, 1H, ArH), 7.80 - 7.81 (m, 1H, ArH).
5,6-Dimethyl-2-phenylbenzoimidazole [43] : m.p. = 251˚C - 252˚C, 1H NMR (300 MHz, DMSO-d6): δ = 2.31 (s, 6H, 2Me), 7.34 - 7.54 (m, 4H, ArH), 8.12 (d, J = 8.0 Hz, 2H, ArH), 12.69 (br s, 1H, NH).
2-(3-Methoxyphenyl)-5,6-dimethylbenzoimidazole [43] : m.p. = 240˚C - 243˚C. 1H NMR (300 MHz, DMSO-d6): δ = 2.31 (s, 6H, 2Me), 3.84 (s, 3H, OMe), 6.99 - 7.03 (m, 2H, ArH), 7.30 - 7.45 (m, 2H, ArH), 7.69 - 7.72 (m, 2H, ArH), 12.60 (1, br s, NH).
NOTES
![]()
*Corresponding author.