Refined Agent Model Using Shape-Based Grained Structure: Application to Membrane Molecules ()
1. Introduction
In this paper, we will first describe the constraints of modeling systems of biological molecules in Section 2. Then we will describe the Multi-Agent System we designed in Section 3. And we will discuss about computational constraints in Section 4.
2. Modeling Molecular Interactions
In a metabolic system, molecule interactions are defined firstly by chemical interactions given by their atom composition (attractive and repulsive forces) and secondly by biochemical relationships such as specific enzyme/substrate reactions. For years, models have been made for each of these interactions separately: molecular dynamics is used for atom-atom interactions [3] or computational models as agent-based systems for enzymatic systems [4] [5] . In this project, we are interested in modeling mitochondria and more particularly, the respiratory chain, a metabolic pathway (enzymatic reaction chain) which takes place inside the inner mitochondrial membrane. This metabolic pathway involves protein complexes moving through the membrane to exchange electrons. Looking at the respiratory chain the necessity of bringing different approaches together emerges because spatial distribution of proteins, mechanical interactions and enzymatic reactions need to be modeled to make sure that mechanical constraints are considered in the process.
2.1. Biological Membranes
A biological membrane is mainly composed of lipids and proteins (Figure 1 illustrates
this composition). Lipids are organized as a bilayer with associated proteins (peripheral proteins) or inserted proteins (transmembrane proteins). Chemically, a membrane can be separated in a hydrophobic leaflet surrounded by two hydrophilic leaflets. This structure creates frontiers between living cells and the outside or defines organelles in a cell, for instance the mitochondrial membrane. Another point is that transmembrane proteins must have a specific chemical composition to stay in the membrane but have exposed sections on the outside whereas peripheral proteins are mostly hydrophilic with a hydrophobic anchor in the membrane. So to model biological reactions in a membrane at the molecular level, we need a way to simulate lipids and proteins with chemical interactions for movements and reaction rules for the metabolic pathway.
2.2. Simulation of Biological Molecules
Molecular dynamics (MD) models have been developed for lipids and membranes (see [6] for a description of the main characteristics). It is a class of computational method to simulate molecules by calculating the movement of each atom in the system. These kinds of techniques is limited by the short duration of simulation (picoseconds, femtosecond) and the small system size it can compute due to the number of atoms and interactions to compute. As the number of lipid molecules can be high leading to a high number of atoms to simulate, these models usually simplify lipid structures using grains [7] [8] . We have based our lipid description on a previous work of Farago [9] which models them as a rigid molecule composed of three grains. The equation details will be given in Section 3.3.
2.3. Modeling Protein Complexes
One more constraint for modeling proteins is that internal movements are important for enzymatic reactions [12] . Proteins are flexible molecules and they have specific sites where these reactions can happen. Sometimes sites can be accessible only if the protein is in the good conformation. In the respiratory chain metabolic pathway, proteins exchange electrons to produce energy for the cell. The distance between binding sites is especially important as it can lead to short-circuits and a loss of efficiency.
Flexible parts can be identified comparing alternative positions of atoms. The Hingefind algorithm [13] proceeds to a superposition of two structures of a same molecule in different states to find large domains of atoms which move together. A drawback of this approach is that this algorithm needs two different structures and data are not always available. This lack of data can be solved by using normal modes. Normal Modes Analysis (NMA) or Elastic Network Models (ENM) consider a protein as a set of harmonic oscillators and they allow to determine possible positions of the protein atoms.
Using these methods, we have determined a set of Constraint Rigid Bodies (CRB) for Complex II and Complex III of the repiratory chain. Complex II can be divided in 2 CRB (see Figure 2): the most important is the external part (in blue) and the other one is the transmembrane part (in red). Complex III can be divided in three CRB [14] (see Figure 3): the main body is composed of the transmembrane part and the matrix part
(in red) but there are two other small bodies (in blue) which correspond to the extrinsic part of the iron-sulfur protein (ISP) of this complex. We called them the ISP heads. These articulated domains are located in the intermembrane space and that the transmembrane part has no articulation inside the membrane. We know angles and directions of rotation as they are results from the Hinge Find algorithm. Combined with the lipid model with three grains, we used this representation of proteins in a multi-agent system to model biochemical interactions.
3. Design of Multi-Agent Model for Molecules
Traditionally, two kinds of agents are considered: reactive and cognitive. Agents to simulate biological entities are usually reactive because molecules can be considered as autonomous but do not have intentions or take decisions. They interact together by applying biochemical or biophysical rules, not because they build plan to achieve a task as cognitive agent. Their spatial environment, the cell, is in 3D and the temporal scale has to match with the biochemical rules which are defined with enzyme kinetics, i.e. velocity of reactions.
3.1. Multi-Agent System Features
To model the different components of the membrane, the authors designed a data structure by using object oriented paradigm (see the class diagram in Figure 4). They considered two classes of agents: ActiveAgents have rules to interact with the environment or with another agents, PassiveAgents just randomly move. Each agent has a Body attribute containing a description of physical properties for a molecule and the agent spatial position. From this generic class, we derived two types of body: Active and Passive, and one subclass of active body which is GrainBody. This is the place where Grain types can be defined: HeadGrain, InterfaceGrain and TailGrain, in the case of our three grain model. Of course other coarse-grain models can be added for other concerns. ActiveBody and PassiveBody are characteristics of active and passive agents respectively.
For example, a lipid molecule is composed of an ActiveBody with a grain of each type embedded in a ThreeGrain body object. Interactions and movements are computed with each grain co-ordinates and radius using special formulas detailed in the next sections.
3.2. Model Environment
The environment is a grid in a 3D space and also stores information that are being used by all agents. Each Grain in an agent belongs to exactly one grid cell. In the case of a lipid agent made of three grains, this agent occupies three cubes. By this way, we can find exactly the neighbors of a grain and compute interactions easily. When a lipid agent is added to the system, it firstly obtains the position of its interface grain in the environment. Then, it receives two free orthogonal positions. The process to find these three positions can be described below. Suppose the position of the interface grain is 13 (see
Figure 5), then its neighbors are marked from 0 to 26. We can choose one of these pairs to assign to head and tail grain: [1 - 25], [4 - 22], [7 - 19], [10 - 16], [12 - 14], [0 - 26], [9 - 17], [18 - 8], [21 - 5], [24 - 2], [15 - 11], [6 - 20], [3, 23].
3.3. Biochemical Interactions
As we have mentioned before, the retained model is a three grain model, which have been previously described by Farago [9] . Figure 6 resumes interactions that can be applied depending on the grain types: number 1 labels the head grain, number 2 the interface and number 3 the tail. Grain 1 is hydrophilic and both 2 and 3 hydrophobic. The biochemical interactions between grains are computed with adapted Lennard- Jones potentials. For each grain in the agent body, the force U is calculated by considering the interactions with its neighbors. In the Farago model, a different equation is applied depending on the type of the grain but after some verification, we have pointed
![]()
Figure 5. Neighbor positions for a cube.
out that it is possible to keep the same equation for all interaction cases with a right set of  and σ values. With two grains i, j of any type, we have:
and σ values. With two grains i, j of any type, we have:

where r is the distance between grains and rc is the cut-off distance. Values of 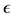 and σ are chosen from Table 1. Distance between grains is important because a grain can only affect another one if it is close enough regarding the cut-off distance. That means that we do not need to compute interaction forces between all the agents in the environment but only those ones inside a limited area.
and σ are chosen from Table 1. Distance between grains is important because a grain can only affect another one if it is close enough regarding the cut-off distance. That means that we do not need to compute interaction forces between all the agents in the environment but only those ones inside a limited area.
Note that the set of neighbors of a grain does not include the other grains in the same agent (see Figure 6). So for a lipid agent composed of three grains, we have three forces: UHead, UInterface, and UTail. Then, the force that affects an agent is:
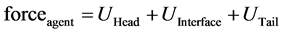
Based on this force, a grain computes its new position by using the equations below:
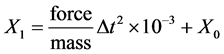
![]()
Table 1. Espilon 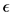 table and sigma σ table for grain interactions.
table and sigma σ table for grain interactions.
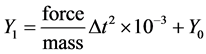

where force is in Newton (kg/m/s2) and mass in Yoto kilogram (1024 kilogram). In our model, mass value is 2.
3.4. Membrane Arrangement
We have set a bilayer of lipids and inserted in the two complexes II and III (see Figure 7). The size of grains has been chosen to recreate the known size of molecules. A membrane lipid is about 30 Å long, so the radius of a grain for a lipid is 5 Å. In order to keep an acceptable computing time, we wanted to model proteins with as few grains as possible. According to Hinge Find results, transmembrane part of proteins are composed of only one CRB but the size of this grain is huge compared to the size of grain modeling lipids. In that case, computing physical interactions between two beads so different in size is difficult to obtain. So, we preferred to split the CRB in two grains easier to integrate in the membrane. Consequently, we have extended the three grain model of lipids to a protein model with three grains, the difference remains in the size of the proteins. The radius of a membrane protein grain is 15 Å, three times the size of the lipid grain. The hinges found with Hinge Find give the rotation possibilities for extrinsic grains. Each protein coarse-grain contains binding sites to capture metabolites.
Depending on information extracted from MD modeling, binding sites are set inside grains in the right geometric position. Protein agent rules define which metabolites they are able to capture and how they run the transformations. For complexes CII and CIII are involved in the ubiquinone cycle and their binding sites exchange electrons to reduce or oxidize the ubiquinone metabolites. A more complete description of the mechanism could be found at [15] .
To stay realistic and observe the behavior of a piece of mitochondrial membrane, we have to initialize a simulation with several thousands of passive agents, hundreds of active lipid agents and tens of active protein agents.
3.5. Computing Constraints
Multi-agent systems are often considered as non-realistic because if each molecule is an agent, observe all agents at each running step is time consuming when you have thousands or millions of agents. First, using Farago model allows us to have implicit water,
![]()
Figure 7. Schematic side view of lipids and proteins organization.
thus molecules of water do not need to be agentified. Second, metabolites which are the most abundant but are simple agents compared to lipids and proteins. In addition, they are changed by proteins and do not execute any action themselves. Therefore we model them as passive agent with one grain and only one rule: randomly moving.
Computational performances are often the main drawback for MAS. The needs to implement thousands of agents and the multiple direct interactions between agents remain the main cost in computing time, comparing for example with an ODE system. In general to solve this issue, computer scientists turn to parallel programming to find optimization solutions. Unfortunately e, the classical way to parallel program, i.e. CPU parallelization, is not really efficient for agent models. This method consists in splitting data between several kernels, computing them separately and merging the results at the end. As each action of an agent could be observed by all the others at each running step, it is necessary to share information between agents and the cost of this sharing is so high that the benefit is erased. More recently, a solution has arisen with the new way to parallel algorithms, GPU programming which allows to execute peace of code on different kernel but kernels can share one memory space. With this paradigm, agent environment, the grid, can be stored into the shared memory space. Each agent can read information directly into this space and write its own information in the environment. No need to direct communication between agents and the simulations can benefit totally of the agent wide-spread through the kernels. For instance, if the computer uses a graphic card with 1024 kernels, at each run step this number of agents can act at the same time. This method can simulate larger biological model and thus to reach realistic modeling scale.
4. Conclusions
Biological processes involve different types of molecules with complex interactions coming from biophysical and biochemical rules. They take place in 3D environment and the number of entities to include in simulations can be high. This work addresses modeling membrane, more precisely mitochondrial membrane. To do that we have defined two subclasses of reactive agents: active agents and passive agents. Our agents have a body. This body is defined as a grain body in the case of active agents, which are lipids and proteins. It contains all biochemical rules for interactions between agents and enzymatic rules to apply if they capture metabolites. Metabolites are passive agents, meaning that they have a very simple body with only spatial positions and a way to move randomly into the environment.
To model a realistic piece of membrane, the number of required agents is more than one thousand. To optimize simulation time, we have chosen to parallel it by using GPU parallelization. That allows us to execute all agents in only one cycle by spreading them on kernels. From now, it remains to adjust attraction/repulsion parameters between “small” lipid grains and “big” protein grains. But more than to obtain precise results, we have been interested to conceive a tool to simulate complex biological organizations as membranes in order to be able to experiment and to test in silico different behaviors when constraints change. MAS are adequate to achieve this goal. Designing biological processes by individual behavior of molecules is easier than trying to describe a global one at the membrane level. New types of computing method as GPU parallelization open new perspectives to MAS and lead them to become powerful in the domain of biological simulations.