Palladium/Imadazolium Salt Mediated Cyclisations for the Synthesis of Heterocyclic Compounds ()
Received 11 August 2015; accepted 29 November 2015; published 2 December 2015


1. Introduction
Palladium mediated reactions play significant role in organic synthesis. Intramolecular Heck reactions in particular have become useful for the synthesis of carbocylic and heterocyclic rings [1] [2] . Conventional coupling methods require the use palladium phosphine complexes as catalyst for Heck reactions. The mechanism for these palladium catalyzed coupling reactions involves four key stages namely, i) oxidative addition of aryl halide to Pd(0); ii) intramolecular carbometallation; iii) β-hydride elimination and lastly iv) reductive elimination to regenerate the Pd(0) catalyst for the continuation of the catalytic cycle [3] .
The choice of ligand is very important in these palladium catalyzed reactions. In recent times, the development of novel ligands for transition metal catalysis and the study of their mechanistic pathway have received greater attention [4] [5] . Sterically hindered phosphine ligands for palladium-catalysed reactions in particular, have improved greatly the applications of many metal mediated transformations [6] .
Despite their wider application in palladium mediated synthesis, phosphine ligands do suffer limitations. This has necessitated the search for practical alternatives to phosphines as ligands in metal mediated cyclisation reactions [7] .
N-heterocyclic carbenes (NHC) are now potential candidates as alternative ligands in transition metal catalyzed reactions due to their advantages over their conventional phosphine counterparts. They are known to be excellent donors and tend to form strong bonds with metals, giving them very good catalytic activity [7] [8] .
NHCs have also found use in palladium catalysis. In complexes where NHC is coordinated to palladium (II), catalytic activity of these species have been examined and confirmed. In particular, catalytic properties of palladium-carbene complexes have been proven in amination, Heck and Suzuki reactions [9] .
Hartwig and Nolan [10] [11] , have reported that palladium-NHC complexes may be produced as intermediates when imidazolium salts are employed as additives in palladium catalyzed transformations. Although a full mechanistic rational is yet to be established, it is assumed that, the palladium and imidazolium salts generate either neutral or cationic palladium-carbene complexes in situ during the course of the metal catalytic cycle [12] . Nevertheless it can be appreciated that palladium-NHC complexes are proving valuable in synthetic organic chemistry [13] .
We previously reported the successful use of Pd-carbene complexes in intramolecular Heck reactions involving aromatic chlorides. This indicates the effectiveness of these Pd-NHC complexes in metal catalyzed reactions, particularly involving the less reactive aryl chlorides, [14] . Improved yields were achieved with addition of tetrabutyl ammonium salts. These reactions require generation of the carbene in situ from imidazolium salts and subsequent completion of the catalytic cycle leading to the generation of heterocyclic compounds.
We describe herein our findings on palladium/imidazolium salt mediated protocols for intramolecular cyclisation reactions on to aryl iodides. Our studies have revealed that palladium/imidazolium salt catalysis can be used to synthesize a range of heterocyclic compounds from cheaper and readily available starting materials and precursors. Yields of these reactions are particularly good, and the products are free from contamination by phosphine related by-products, making them easily isolable.
2. Experimental
2.1. Chemicals and Instruments
Melting points were carried out on Gallenkemp melting point apparatus and are uncorrected. IR spectra were recorded on a Perkin Elmer transform instrument as thin film neat of a solution dissolved in nujol. All nmr spectra were recorded on Bruker Fourier transform instruments with frequencies quoted in mega Hertz recorded in all experimental data. All coupling constants (J values) were quoted in Hertz. Chemical shifts are reported upfields in parts per million. Mass spectra were recorded on Kratos or Fisons double focusing spectrometers. X-ray crystallography experiments were carried out using Enraf Nonius diffractometer. Thin layer chromatography (tlc) was performed using Merck Kieselgel precoated silica gel plates and visualized with UV light, vaporized iodine or potassium permanganate solution. Flash chromatography was carried out using Merck Kieselgel 230-400 mesh. Organic solvents were purified by distilling over drying agents. Throughout the experimental, imidazolium salt refers to the ligand 1,3-bis-(2,6-diisopropylpheynyl)imidazolium chloride.
2.2. General Method for the Alkylation of Benzyl Alcohol
A suspension of 2-iodobenzylalcohol (1.0 eq) and NaH (2.0 eq) in THF was stirred at room temperature for 30 min. Alkenyl halide (2.0 eq) was added and the reaction mixture stirred for a further 16 h. The reaction mixture was then filtered through celite, washed with water and the aqueous layer extracted with Et2O. The combined organic fractions were dried (MgSO4), filtered and solvent removed in vacuo to give the product as an oil.
2.3. General Method for the Synthesis of Phenylacrylamide
To a solution of acyl chloride (1.0 eq) in Et2O at 0˚C was added slowly 2-iodoaniline (2.0 eq) and the reaction mixture left to warm to room temperature for 2 h. This was then quenched with HCl (0.5 M), washed with NaHCO3 solution followed by water and the aqueous layer extracted with Et2O. The combined organic fractions were dried (MgSO4), filtered and solvent removed in vacuo to give a crude sample which was purified by flash chromatography (Silica gel) [eluent-Pet:Et2O, 20:1 - 5:1] to give the title compound as a solid.
2.4. General Method for the Synthesis of Isochromene and Isochroman
Iodobenzyl ether (1.0 eq), Cs2CO3 (1.5 eq), Pd2(dba)3 (1 mol%) and imidazolium salt (1 mol%) were placed in a reaction vessel and purged with nitrogen under vacuum. N,N-dimethylacetamide was then added and the mixture heated at 140˚C for 15 h. The reaction mixture was allowed to cool to room temperature, dissolved in diethyl ether and filtered through celite. The filtrate was washed with water and aqueous layer extracted with diethyl ether. Combined organic fractions were dried (MgSO4), filtered and solvent removed to give crude sample which was purified by flash chromatography (silica gel) [eluent-Pet:Et2O, 20:1 - 10:1] to give the desired compound.
2.5. General Method for the Synthesis of Benzoxazole
Iodo-benzamide (1.0 eq), Cs2CO3 (1.5 eq), Pd2(dba)3 (1 mol%), and imidazolium salt (1 mol%), were place in a reaction vessel and purged with nitrogen under vacuum. N,N-dimethylacetamide was then added and the mixture heated at 140˚C for 10 h. This was allowed to cool to room temperature, dissolved in diethyl ether and filtered through celite. The filtrate was washed with water and the aqueous layer extracted with ether. Combined organic fractions were dried (MgSO4), filtered and solvent removed to give crude sample which was purified by flash chromatography (silica gel) [eluent-Pet:Et2O, 20:1 - 5:1] to give the desired compound.
3. Spectral and Analytical Data
1-Allyloxymethyl-2-iodobenzene (3.1)
Rf Pet:EtOAc, 6:1 (0.8).
νmaxcm−1 3406, 3048, 2935, 1695, 1451, 1345, 1090, 1004, 915, 736.
δH (300 MHz, CDCl3) 7.72 (1H d, J = 7.7, ArH ), 7.40 (1H, d 7.7, ArH), 7.22 (1H, dd, J = 7.7, 7.3 ArH) 6.87 (1H, app. t, J = 7.7, ArH), 5.90 (1H, ddt J = 17.1, 10.3, 6.7, -CH2OCH2CHCH2), 5.28 (1H, d J = 10.3, -CH2OCH2CHCH2), 5.12, (1H, J = 17.1, -CH2OCH2CHCH2), 4.4 (2H, s, -CH2OCH2CHCH2) 4.02 (2H,d J = 6.7, -CH2OCH2CHCH2).
δC (75 MHz, CDCl3) 141.04 (C), 139.53 (CH), 134.13 (CH), 130.66 (CH), 129.87 (CH), 128.84 (CH), 117.76 (CH2), 98.15 (C), 72.07 (CH2), 69.55 (CH2).
m/z (EI) 274 (M+, 25%), 231 (M+-C3H5, 63%) 217 (100%), 90 (75%).
1-But-2-enyloxymethyl-2-iodobenzene (3.2)
Rf Pet:Et2O, 5:1 (0.88).
νmaxcm−1 3021, 2920, 1562, 1438, 1350, 1092, 1090, 1005, 959, 743.
δH (300 MHz, CDCl3) 7.70 (1H d, J = 7.9, ArH ), 7.33 (1H, d 8.2, ArH), 7.22 (1H, dd, J = 7.7, 7.3 ArH) 6.83 (1H, dd, J = 7.7, 7.3 ArH), 5.69 (1H, dq J = 15.2, 6.0, -CH2OCH2CHCHCH3), 5.58 (1H, dt J = 15.2, 6.0 -CH2OCH2CHCHCH3), 4.36, (2H, s, -CH2OCH2CHCH3), 3.39 (2H, d, J = 5.8, (-CH2OCH2CHCHCH3), 1.63 (3H, d J = 6.0, -CH2OCH2CHCHCH3).
δC (75 MHz, CDCl3) 141.15 (C), 139.51 (CH), 130.25 (CH), 129.16 (CH), 128.72 (CH), 127.81N(CH), 127.08 (CH), 98.26 (C), 71.84 (C), 66.43 (CH2), 18.33 (CH3).
m/z (EI) 288 (M+, 20%), 244 (M+, 40%) 217 (M+-OC4H7, 100%), 36 (60%).
1-Cynnamyloxymethyl-2-iodobenzene (3.3)
Rf Pet:Et2O, 9:1 (0.55).
νmaxcm−1 3033, 2848, 1557, 1443, 1354, 1107, 1090, 960, 746, 687.
δH (300 MHz, CDCl3) 7.92 (1H d, J = 7.5, ArH ), 7.60 (1H, d J = 7.9, ArH), 7.42 (2H, dd, J = 7.5, 7.4 ArH), 7.32-7.10 (4H, m, ArH), 7.07 (1H, apt. t J = 7.9 ArH), 6.57 (1H, d, J = 16.0, -CH2OCH2CHCHPh), 4.65 (2H, s, -CH2OCH2CHCHPh), 4.37 (2H, d J = 6.0, -CH2OCH2CHCHPh).
δC (75 MHz, CDCl3) 141.108 (C), 139.68 (CH), 137.18 (C), 133.13 (CH), 129.71 (CH), 129.34 (CH), 129.10 (CH), 128.76 (CH), 128.25 (CH), 127.07 (CH), 126.35 (CH), 98.47 (C), 76.43 (CH2), 71.79 (CH2).
m/z (ES) 386 (MNH4+, 100%), 305 (100%).
N-(2-Iodophenyl)-3-phenylacrylamide (3.6)
Rf Pet:EtOAc 6:1 (0.41).
Mp. 160˚C - 162˚C.
νmaxcm−1 3189, 1655, 1620, 1451, 1374, 731.
δH (300 MHz, CDCl3) 8.3 (1H, appt. d J = 7.5, NH), 7.74-7.72 (1H, m, ArH), 7.68 (1H, d J = 15.8, -COCHCH-), 7.56-7.49 (3H, m, ArH), 7.36-7.18 (4H, m, ArH), 6.79 (1H, appt. t J = 7.9, ArH), 6.52 (1H, d J = 15.5 -COCHCH-).
δC (75 MHz, CDCl3) 164.30 (C=O), 143.41 (C), 139.24 (C), 138.70 (CH), 134.82 (CH), 130.60 (CH), 129.75 (CH), 129.32 (CH), 128.50 (CH), 126.44 (CH), 122.46 (CH), 120.98 (CH), 90.05 (C).
m/z (EI) 349 (M+, 30%), 222 (M+-I, 80%), 131 (M+-C6H5NI, 100%).
N-(2-Iodophenyl)-benzamide (3.7)
Rf Pet:Et2O 6:1 (0.39).
Mp. 140˚C - 142˚C.
νmaxcm−1 3214, 1639, 1506, 1456, 1373, 1287, 739, 704.
δH (300 MHz, CDCl3) 8.40 (1H, d J = 8.2, ArH), 8.22 (1H, s, NH), 7.90 (2H, dd, J = 8.2, 7.9, ArH), 7.74 (1H, d J = 7.9, ArH), 7.56-7.40 (3H, m, ArH), 7.34 (1H, dd, J = 8.2, 7.3, ArH), 6.81 (1H, dd, J = 7.5, 7.7, ArH).
δC (75 MHz, CDCl3) 165.70 (C=O), 139.21 (C), 138.66 (C), 134.92 (CH), 132.60 (CH), 130.57 (CH), 129.83 (CH), 129.36 (CH), 127.57 (CH), 126.44 (CH), 122.14 (CH), 90.59 (C).
m/z (EI) 323 (M+, 10%), 196 (M+-I, 62%), 122 (87%), 105 (M+-C6H5NI, 100%).
4-Methyl-1H-isochromene (4.1)
Rf Pet:Et2O, 9:1 (0.87).
νmaxcm−1 3048, 2844, 1640, 1484, 1354, 1448, 1140, 1107, 1007, 936, 72.
δH (300 MHz, CDCl3) 7.18, (1H d, J = 7.5, ArH ), 7.07 (1H dd J = 7.5, 7.4 ArH), 7.01 (1H, d, J = 7.5 ArH), 6.93 (1H, dd J = 7.5, 7.3, ArH), 6.39 (1H, s, -CH2OCHC-Me), 4.91 (2H, s, -CH2OCH-), 1.84 (3H, s, CH3).
δC (75 MHz, CDCl3) 141.28 (CH), 131.34 (CH), 127.68 (C), 127.02 (CH), 125.61 (CH), 122.68 (CH), 119.26 (CH), 110.36 (C), 67.20 (CH2), 12.07 (CH3).
m/z (EI) 146 (M+, 85%), 117 (100%), 91 (15%).
4-Ethyl-1H-isochromene (4.2)
Rf Pet:Et2O, 10:1 (0.88).
νmaxcm−1 3062, 2960, 2834, 1628, 1483, 1448, 1139, 1107, 935, 747.
δH (300 MHz, CDCl3) 7.18, (1H d, J = 7.0, ArH ), 7.09 (2H dd J = 7.7, 7.3 ArH), 6.95 (1H, d, J = 7.9 ArH), 6.40 (1H, s, -CH2OCHCCH2CH3), 4.89 (2H, s, -CH2OCHCCH2CH3), 2.31 (2H, q J = 7.5, -CH2OCHCCH2CH3) 1.09 (3H, t J = 7.5, -CH2OCHCCH2CH3).
δC (75 MHz, CDCl3) 142.26 (CH), 131.92 (2XC), 129.41 (CH), 128.39 (CH), 126.87 (CH), 124.36 (CH), 120.58 (CH), 118.10 (C), 68.65 (CH2), 21.14 (CH2), 13.92 (CH3).
m/z (EI) 160 (M+, 85%), 117 (100%), 91 (55%), 49 (80%).
4-Benzyl-1H-isochromene (4.3)
Rf Pet:Et2O 9:1 (0.8).
Mp. 53˚C - 55˚C.
νmaxcm−1 29684, 1623, 1454, 1448, 1141, 956.
δH (300 MHz, CDCl3) 7.27, (1H d, J = 7.0, ArH ), 7.22-7.19 (3H, m, ArH), 7.08-7.05 (3H, m, ArH), 6.95 (2H, appt. t, J = 7.7 ArH), 6.40 (1H, s, -CH2OCHC-), 4.96 (2H, s, -CH2OCHCC-), 3.61 (2H, s-CH2Ph).
δC (75 MHz, CDCl3) 144.79 (CH), 140.02 (C), 131.74 (2XC), 129.16 (CH), 128.81 (2 × CH), 128.78 (CH), 128.42 (CH), 127.07 (CH), 126.58 (CH), 124.29 (CH), 121.31 (CH), 115.10 (C), 68.81 (CH2), 34.36 (CH2).
m/z (EI) 222 (M+, 55%), 57 (57%), 43 (100%), 28 (60%).
4-Benzylidene-isochroman (4.4)
Rf Pet:Et2O 9:1 (0.5).
Mp. 78˚C - 80˚C.
Elemental Analysis: Expected; C 86.45%, H 6.35%, O 7.20%.
Found; C 86.62%, H 6.24%, O 7.14.
νmaxcm−1 3379, 2983, 1712, 1596, 1492, 1447, 1268, 1100, 764.
δH (300 MHz, CDCl3) 7.66 (1H, d J = 7.3, ArH), 7.27, (2H appt t, J = 7.5, ArH), 7.19 - 7.12 (5H, m, ArH), 7.08 (1H, s, -CH2OCH2CCH-Ph), ArH), 6.95 (1H, d, J = 7.3 ArH), 4.68 (2H, s, -CH2OCH2CCH-Ph), 4.66 (2H, d J = 1.5 -CH2OCH2CCH-Ph).
δC (75 MHz, CDCl3) 137.09 (C), 135.27 (C), 132.68 (C), 132.57 (C), 129.72 (CH), 128.78 (CH), 128.04 (CH), 127.60 (2 × CH), 127.59 (2 × CH), 126.58 (CH), 125.15 (CH), 123.81 (CH), 123.66 (CH), 69.01 (CH2), 67.28 (CH2).
m/z (EI) 222 (M+, 100%), 178 (60%), 115 (95%).
2-Styryl-benzooxazole (4.5)
Rf Pet:EtOAc 3:1 (0.7).
Mp. 90˚C - 92˚C.
νmaxcm−1 2984, 1637, 1530, 1453, 1372.
δH (300 MHz, CDCl3) 7.81 (1H, d J = 16.3, -CHCHPh), 7.74 (1H, d J = 7.5 ArH) 7.73 (2H, dd J = 7.6, 7.4, ArH), 7.54 (1H, d J = 7.5 ArH), 7.43 (2H, dd J = 7.6, 7.6 ArH), 7.36-7.34 (3H, m ArH), 7.10 (1H, d J = 16.3 - CHCHPh).
δC (75 MHz, CDCl3) 161.76 (-NC), 149.37 (C), 141.15 (C), 138.42 (CH), 134.1 (C), 128.74 (CH), 127.94 (CH), 127.33 (CH), 126.52 (CH), 126.20 (CH), 124.18 (CH), 123.47 (CH), 118.84 (CH), 112.91 (CH), 101.29 (CH).
m/z (EI) 221 (M+, 80%), 220 (M+ - H 100%), 191 (45%).
2-Phenyl-benzooxazole (4.6)
Rf Pet:EtOAc 6:1 (0.81).
Mp. 110˚C - 112˚C.
νmaxcm−1 2986, 1611, 1550, 1458, 1373, 1233, 1045, 731.
δH (300 MHz, CDCl3) 8.22-8.17, (2H, m, ArH), 7.72-7.69 (1H, m, ArH), 7.47-7.44 (4H, m, ArH), 7.30-7.27 (2H, m, ArH).
δC (75 MHz, CDCl3) 149.73 (C), 141.07 (C), 130.49 (C), 127.89 (2 × CH), 126.59 (2 × CH), 126.13(C), 126.20 (CH), 124.08 (CH), 123.55 (CH), 118.99 (CH), 109.57 (CH).
m/z (EI) 195 (M+, 100%), 167 (50%), 77 (40%), 63 (50%).
4. Results and Discussion
Starting materials were obtained by standard synthetic transformations, where 2-iodobenzylalcohol was deprotonated and the anion subsequently quenched with the corresponding alkenylbromide [15] [16] , to furnish the desired alkenyloxymethyl-2-iodobenzene as the cyclisation precursors, 3.1-3.3 (Scheme 1). To obtain oxyindoles as final heterocyclic products, acrylamide precursors were synthesized by direct acylation of iodoaniline with enoyl chlorides to give desired products 3.4-3.6 (Scheme 2), [17] .
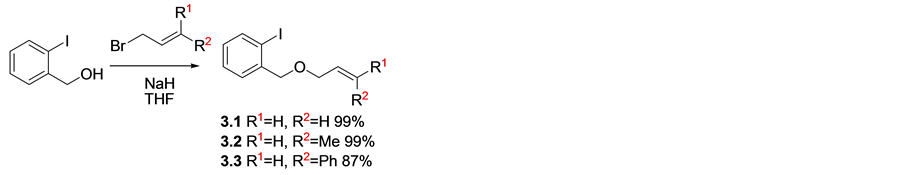
Scheme 1. Synthesis of benzyloxy precursors used in the synthesis of bicyclic rings.

Scheme 2. Synthesis of amide precursors which gave oxazoles upon cyclisations.
When alkenyloxy precursors 3.1 and 3.2 were subjected to reaction conditions using the palladium/imidazo- lium salt protocol, the desired bicyclic isochromene products 4.1 and 4.2 were isolated in very good yields following purification by column chromatography. The cyclisations proceeded possibly via 6-exo fashion followed by rapid isomerization to give the products obtained, (Scheme 3).
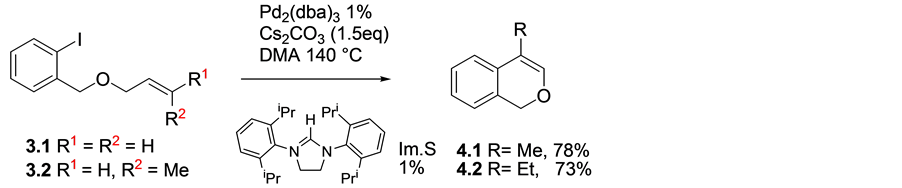
Scheme 3. Pd/ Im.S mediated cyclisations to produce bicyclic ring systems (Im.S = imidazolium salt).
When the phenyl substituted precursor 3.3 was subjected to the same reaction conditions, however, two isomeric products were isolated [18] . Benzylisochromene 4.3 [19] , and benzylideneisochroman 4.4 [20] [21] , nuclei were obtained in yields of 22% and 63% respectively, (Scheme 4).
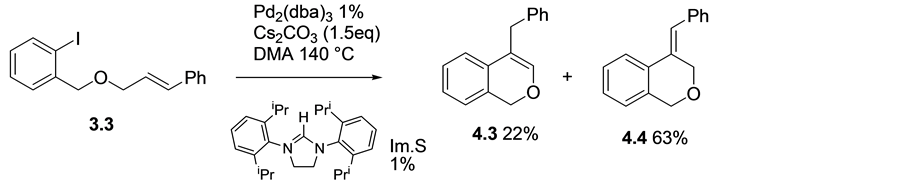
Scheme 4. Phenyl substituted precursor gave isomers 4.3 and 4.4 when subjected to Pd/ Im.S conditions.
Isolation of benzylideneisochroman 4.4 as the major product in this case means the reaction probably proceeded via 6-exo cyclisation. Unlike the previous examples where isomerization is presumed to have taken place following cyclisation to give the kinetically stable products, benzylideneisochroman 4.4 (Figure 1) happen to be the major isolated product from cyclisation of 1-cynnamyloxymethyl-2-iodobenzene, 3.3. Although there is a possible 1,3-hydrogen shift to generate isochromene 4.3, the isolation of isochroman 4.4 as the major product may be due to the conjugation system that extends from the alkene bond onto the phenyl ring to give a more stable isomer.
![]()
Figure 1. Crystal structure of benzylideneisochroman 4.4, major isomer from phenyl substituted precursor.
It was disappointing to observe that, treating but-2-enoic phenyl amide and phenyl acrylamide substrates with palladium/imidazolium salt failed to give the desired oxyindoles. Surprisingly, what seemed to be loss of the al- kenoyl moiety leading to isolation of 2-iodoaniline was observed, (Scheme 5).
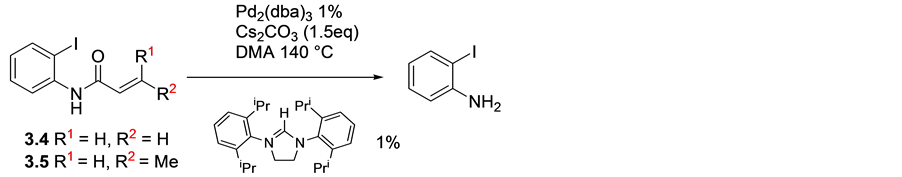
Scheme 5. Reaction of alkenoyl moiety upon treatment with Pd/Im.S.
When N-(2-iodophenyl)-3-phenylacylamide 3.6 was subjected to palladium/imidazolium salt protocols, there was conversion of starting material to product after heating for 3 h. It was revealed by spectral analysis that the desired oxyindole had not been formed, instead cyclisation had occur to give 2-styryl-benzooxazole 4.5, [22] [23] , (Scheme 6), (Figure 2) [24] .
![]()
Scheme 6. Cyclisation to give 2-styryl-benzooxazole using Pd/Im.S.
![]()
Figure 2. Crystal structure of 2-styryl-benzooxazole, 4.5.
To further extend the scope of these findings, N-(2-iodophenyl)benzamide 3.7 and N-(2-iodophenyl) aceta- mide were subjected to the same reaction conditions. While the benzamide reacted possibly via o-arylation to produce 2-phenylbenzooxazole 4.6 in good yield, no product was isolated when the acetamide derivative was subjected to the same reaction conditions, (Scheme 7).
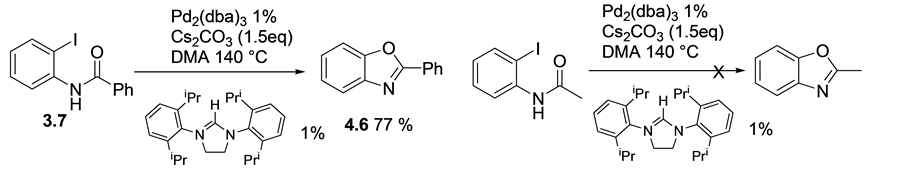
Scheme 7. Under Pd/Im.S condition, iodophenylbenzamide gave Phenyl benzooxazole, 4.6 whiles the acetamide intermediate gave no desired product.
We believe the ability of the phenyl substrates to efficiently undergo cyclisation may be due to the phenyl group providing strong steric and electronic effects leading to a stable rotamer with a configuration that allows the carbonyl oxygen to be within close proximity to the iodine, and thus favouring an attack on to the ring, (Scheme 8).
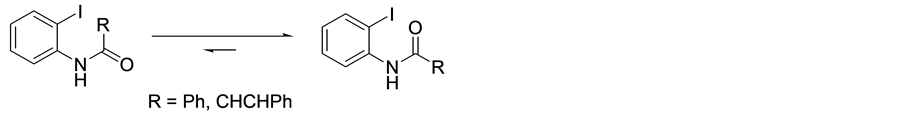
Scheme 8. Rotation around the carbonyl showing a shift in equilibrium to the more favourable rotamer.
5. Conclusion
In conclusion, we have shown that palladium imidazolium salt protocols can be used to promote intramolecular cyclisation reactions, enabling the synthesis of fused bicyclic heteroaromatics with great efficiency. Phenyl substituents have strong influence on reaction outcomes as a result of steric and electronic factors. Iodophenylbenzamide substrates undergo cyclisation to give substituted benzooxazoles. The ease with which phenyl containing amides undergo cyclisation to produce substituted benzooxazoles is noteworthy.
Acknowledgements
We gratefully acknowledge Dr. John Hayler at GSK Tonbridge for his encouragement, support and helpful comments throughout this project. We also gratefully acknowledge support of this project from GSK and EPSRC. We are grateful to EPSRC Mass Spectrometry Service at Swansea and Drs Abdul-Sada, Avent and Hitchcock of The University of Sussex for their contributions to this work. We gratefully acknowledge the support by members of staff of department of pharmaceutical chemistry KNUST, Kumasi Ghana. We are also grateful to Lagray Chemicals, Ghana Limited for their regular support.
NOTES
![]()
*Corresponding author.