Synthesis of Polysilanes Using Mg and Lewis Acid and the Consideration of This Reaction Mechanism ()
1. Introduction
Polysilanes (PS) consisting of silicon-silicon bond chain (Figure 1) has attracted considerable attention due to their usefulness as the precursors for thermally stable ceramics [1] [2] materials for microlithography [3] -[5] , materials for solar cell element [6] and coating materials for PE pipe connection [7] [8] .
In contrast to the growing interest with the PS, the method of preparation hitherto known is highly limited. So far, the almost only practical method is the condensation of dichlorosilanes with alkali metal (the Kipping Method) [9] . This method, however, requires drastic reaction conditions and hence is very much limited as a practical method of PS. Although several modified or alternative methods such as electrochemical coupling of dichlorosilane using Mg electrodes [10] - [14] , transition metal catalyzed reaction of hydrosilane [15] [16] , anionic polymerization of masked disilene [17] - [19] , and ring opening polymerization of cyclic organosilane [20] [21] have been proposed, they are not always extensively effective as the preparative methods (Figure 2).
![]()
Figure 2. Representative preparative method of PS.
On the other hand, it has been reported in our previous studies, the reduction of dichlorosilane with Mg metal in the presence of Lewis acid (MCln) is highly useful method for the synthesis of PS under extremely mild reaction conditions (Scheme 1) [22] - [24] .
In this paper, the details of our method are described including mechanistic view points and we also wish to report that it is successfully applied for the synthesis of various types of PS having a linear structure with high molecular weight, a cyclic structure, a linear structure with low molecular weight that is suitable for coating materials for PE [7] [8] and silane-styrene copolymers as another type of PS.
2. Experimental
2.1. General
1H NMR and 29Si NMR spectra were obtained in JEOL 400 MHz NMR spectrometer (ECX-400), and were recorded in following conditions. Solvent; tetrahydrofuran-d8 (THF-d8), temperature; room temperature, measurement technique; single pulse method (1H), gated proton decoupling method (29Si), accumulation times; 16(1H), 5000(29Si). Field desorption mass spectrometry (FD-MS) were obtained in JEOL double focusing mass spectrometer (JMS-700) and the analysis conditions are as follows, ionization method; field desorption, field emitter; carbon, emitter current; 0 - 40 mA (2 mA/min.), acceleration voltage; 8 kV. Molecular weights of polymers and oligomers were determined by gel-permeation chromatography (GPC) using THF as an eluent, relative to polystyrene standards. Elemental analyses were performed by KRI Co., Ltd. materials analysis research center.
2.2. Poly [Methlphenylsilane]
The reaction was carried out as follows; Into a 30 mL round bottomed flask were added THF (30 ml, dried over Na), Mg powder (40 mmol), LiCl (20 mmol), and ZnCl2 (4 mmol) as a Lewis acid under N2 atmosphere and stirred to solve LiCl and ZnCl2 to THF for an hour. Dichloromethylphenylsilane (1, 30 mmol) was added into the flask, and the mixture was stirred for 24 h at room temperature. After 3 ml of water and toluene (50 ml) was added into the reaction mixture, it was filtered to remove Mg salts. The filtrate was poured into an ice cold solution of HCl (1 M/dm3, 100 mL) and the aqueous solution was extracted with toluene (50 mL × 3). The combined organic layers were washed twice with 50 mL of brine, dried over MgSO4, and concentrated. The products were purified by reprecipitation from IPA (100 ml). The reactions shown in Table 3 and Table 4 were carried out under similar conditions as described above. The product showed reasonable NMR and IR spectra for the assigned structures. UV λmax (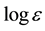 ) (THF) 334.7 nm (3.95); IR (KBr) 3050, 2960, 1430, 1250, 1100 cm−1; 1H NMR (THF-d8) δ −1.00 - 0.70 (m, 3H, SiMe), 6.30 - 7.70 (m, 5H, ring protons), Mn = 8971, Mw = 19,540.
) (THF) 334.7 nm (3.95); IR (KBr) 3050, 2960, 1430, 1250, 1100 cm−1; 1H NMR (THF-d8) δ −1.00 - 0.70 (m, 3H, SiMe), 6.30 - 7.70 (m, 5H, ring protons), Mn = 8971, Mw = 19,540.
2.3. Poly [Diphenylsilane]
As shown in Figure 6, test samples with a width of 1 cm was cut from the joint portion of the PE pipe and electrofusion joint in a direction parallel to the PE pipe. Eight sample pieces were obtained from the PE pipe having a diameter of 50 mm. The test piece was fixed to a tensile machine to determine the maximum peeling load applied at the fusion interface between the PE pipe and electrofusion joint. By elongating the PE pipe, the maximum peeling load was increased up to 700 N, and showed a rapid decrease by peeling at the fusion interface. The weld energy was calculated from the peel load at each elongation distance.
The polymerization of dichlorodiphenylsilane was carried out by the same procedure of dichloromethylphenylsilane. UV λmax (THF) 240 nm; IR (KBr) 3060, 2990, 1430, 1260, 730 cm−1; 1H NMR(THF-d8) δ 6.86 - 6.92 (m, 2H), 7.08 - 7.15 (m, 1H), 7.21 - 7.30 (m, 2H), 13C NMR (THF-d8) δ128.1, 129.2, 135.4, 138.7, Mn = 1000, Mw = 1100.
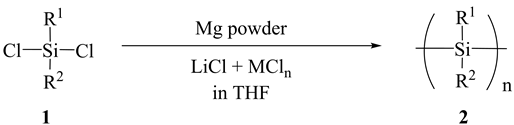
Scheme 1
2.4. Poly [Phenylsilyne]
The polymerization of phenyltrichlorosilane was carried out by the same procedure of dichloromethylphenylsilane. UV λmax (THF) 240 nm; IR (KBr) 3010, 2990, 1490, 1430, 1090, 690 cm−1; 1H NMR (THF-d8) δ 6.30 - 6.70 (m, 5H), Mn = 1380, Mw = 1940.
2.5. Poly [Hexylmethyl-Methylphenylsilane]
The polymerization of the mixture of dichlorohexylmethylsilane (15 mmol) and dichloromethylphenylsilane (15 mmol) was carried out by the same procedure of dichloromethylphenylsilane. UV λmax (THF) 328 nm; IR (KBr) 3050, 2930, 2830, 1430, 1250, 1090 cm−1; 1H NMR (THF-d8) δ −0.90 - 0.65 (m, 8H), 1.10 - 1.75 (m, 11H), 6.40 - 7.35 (m, 5H), Mn = 6200, Mw = 9200.
2.6. Poly [Methlphenylsilane] End-Capped by Trimethylsilyl Groups
The polymerization of the mixture of dichloromethylphenylsilane (25 mmol) and chlorotrimethylsilane (15 mmol) was carried out by the same procedure of dichloromethylphenylsilane. UV λmax (THF) 328 nm; 1H NMR (THF-d8) δ −0.20 - 0.52 (m, 7.5H), 7.10 - 7.60 (m, 5H), Mn = 950, Mw = 1050.
2.7. Poly [Methlphenylsilane]-Polystyrene Random Copolymer (10a)
Into a 30 mL round bottomed flask were added THF (30 ml, dried over Na), Mg powder (40 mmol), LiCl (20 mmol), and ZnCl2 (4 mmol) as a Lewis acid under N2 atmosphere and stirred to solve LiCl and ZnCl2 to THF for an hour. Dichloromethylphenylsilane (1, 15 mmol) and styrene monomer (15 mmol) were added into the flask at the same time, and the mixture was stirred for 24 h at room temperature. After 3 ml of water and toluene (50 ml) was added into the reaction mixture, it was filtered to remove Mg salts. The filtrate was poured into an ice cold solution of HCl (1 M/dm3, 100 mL) and the aqueous solution was extracted with toluene (50 mL × 3). The combined organic layers were washed twice with 50 mL of brine, dried over MgSO4, and concentrated. The products were purified by reprecipitation from IPA (100 mL). A desired polymer was obtained in 71% yield. The molecular weight was determined by GPC based on polystyrene standard. The product showed reasonable 1H NMR, 29Si NMR and IR spectra for the assigned structures.
10a: UV λmax (THF) 328 nm; IR (KBr) 3050, 2920, 2850, 1500,1450, 1250, 1070, 750, 700 cm−1; 1H NMR (THF-d8) δ −1.00 - 0.70 (m, 3H), 1.25 - 1.70 (m, 2H), 1.75 - 2.02 (m, 1H), 6.31 - 7.25 (m, 10H), 29Si NMR (THF-d8) δ −44 - −36 (1.00 Si), ?19 - ?10 (1.02 Si), 2 - 4 (1.09 Si), Mn = 3700, Mw = 4760.
2.8. Poly [Methlphenylsilane]-Polystyrene Block Copolymer (10b)
Into a 30 mL round bottomed flask were added THF (30 ml, dried over Na), Mg powder (40 mmol), LiCl (20 mmol), and ZnCl2 (4 mmol) as a Lewis acid under N2 atmosphere and stirred to solve LiCl and ZnCl2 to THF for an hour. Dichloromethylphenylsilane (1, 15 mmol) was added into the flask, and the mixture was stirred for 3 h at room temperature. After 3 h stirring, styrene monomer (15 mmol) was added into the flask and stirred another 21 h. After 3 ml of water and toluene (50 mL) was added into the reaction mixture, it was filtered to remove Mg salts. The filtrate was poured into an ice cold solution of HCl (1 M/dm3, 100 mL) and the aqueous solution was extracted with toluene (50 mL × 3). The combined organic layers were washed twice with 50 mL of brine, dried over MgSO4, and concentrated. The products were purified by reprecipitation from IPA (100 ml). A desired polymer was obtained in 68% yield. The molecular weight was determined by GPC based on polystyrene standard. The product showed reasonable 1H NMR, 29Si NMR and IR spectra for the assigned structures. 10b: UV λmax (THF) 331 nm; IR (KBr) 3050, 2920, 2850, 1500,1450, 1250, 1070, 750, 700 cm−1; 1H NMR (THF-d8) δ −1.00 - 0.70 (m, 3H), 1.25 - 1.70 (m, 2H), 1.75 - 2.02 (m, 1H), 6.31 - 7.25(m, 10H), 29Si NMR (THF-d8) δ ?44 - ?36 (1.00 Si), −19 - −10 (0.75 Si), 2 - 4 (0.94 Si), Mn = 4100, Mw = 5240.
2.9. Poly [Methlphenylsilane]-Polystyrene Block Copolymer (10c)
Styrene monomer (15 mmol) was added after dichloromethylphenylsilane(1, 15 mmol) was stirred for 7 h. This reaction was carried out by the same procedure of 10b. Desired polymer was obtained in 70% yield. 10c: UV λmax (THF) 332 nm; IR (KBr) 3060, 2920, 2850, 1500,1450, 1250, 1070, 750, 700 cm−1; 1H NMR (THF-d8) δ −1.00 - 0.70 (m, 3H), 1.25 - 1.70 (m, 2H), 1.75 - 2.02 (m, 1H), 6.31 - 7.25(m, 10H), 29Si NMR (THF-d8) δ −44- −36 (1.00Si), −19 - −10 (0.071Si), 2 - −4 (0.037 Si), Mn = 6700, Mw = 16,000.
3. Results and Discussions
3.1. Synthesis of Polymethylphenylsilane Using Mg Metal, LiCl, and Lewis Acid
Mg metal particles, catalysts and THF were added into a three-necked flask equipped with a dropping funnel, a condenser and a mechanical stirrer under a nitrogen atmosphere. After 30 min stirring at 25˚C, dichloromethylphenylsilane (1) (Scheme 1, R1 = Ph, R2 = Me) was dropped into the flask and it was not long before the condensation reaction occurred.
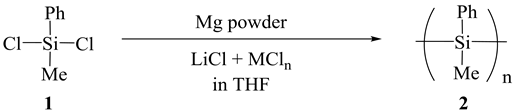
The reaction was quenched by adding IPA, and the mixture was then poured into ice cold 1 N HCl and the aqueous solution was extracted with toluene. The combined organic layer was washed with brine, dried over MgSO4 and concentrated. The resulting crude polymer was dissolved in toluene and purified by reprecipitation from IPA. The molecular weight of the polymer was determined by GPC with THF as the eluent. The reduction of 1 using Mg metal was studied as a model reaction and carried out under a variety of reaction conditions. The results are shown in Table 1 and Figure 3.
First, in case of using only Mg without any catalysts, 1 still remained and polymethylphenylsilane (2) was not obtained. Mg is much less reactive than alkali metal such as Na (ionization potential, Mg = 7.6 eV, Na = 5.1 eV). Second, LiCl was employed as a catalyst. However, it did not dissolve in THF at all and the condensation reaction did not occur. To increase the solubility of LiCl in THF, another metal chloride was added to the reaction mixture. That is to say, it was found that 1 was gradually reduced in the case of using FeCl2 in addition to LiCl as catalysts and 2 was obtained in good yield. Incidentally, the reaction did not also occur using only FeCl2 as a catalyst although it was soluble in THF.
As shown in the Table 2, reaction temperature was important as one of the factors to the formation of Si-Si bond, that is, PS synthesis. The reaction was not initiated at 10˚C (run 1), but occurred at 25˚C and 35˚C (runs 2, 3) accompanied by mild exothermic reactions creating PS. However, the decrease of the yield of PS was observed when the reaction was carried out at 50˚C (run 4). And at 65˚C (run 5, boiling point of THF) the reaction was not initiated. The reaction at high temperature would take place the reduction of FeCl2 with Mg at first, and FeCl2 would be consumed.
The effects of Lewis acid catalysts were shown in Table 3. As shown in the Table 3 (runs 1 - 6), some kinds of Lewis acids were found to be effective for the synthesis of 2, however, the use of AlCl3 resulted in the low yield and low molecular weight of 2. The reason may be explained by the cleavage of Si-Si bonds by the strong Lewis acids (AlCl3) [25] - [27] .
The measurement of UV spectra stemming from Si-Si σ-conjugation indicated the interesting effect of Lewis acid catalysts. Measuring the maximum absorbed wavelength λmax of the obtained 2 in ultraviolet region (Figure 4 ), it was found that 2 prepared by using ZnCl2 as the catalyst had the longest absorbed wavelength (around 335 nm). This showed that 2 synthesized with ZnCl2 had the least structural defects and a straight chain structure of Si-Si bonds.
In addition, the effect of Lewis acid was also observed in the structures of 2. As shown in Figure 5, 1H-NMR of 2a (2 prepared by using FeCl2) and 2b (2 prepared by using ZnCl2) showed similar signals at δ −1.00 - −0.70 (m, Me), and 6.30 - 7.70 (m, Ph), however, exact proportion between methyl protons and ring proton of 2a was 3.3:5.0 and that of 2b was 3.1:5.0 (theoretical value is 3.0:5.0). This means that 2a has more branched Si chains than 2b has. This is also supported by the 29Si-NMR chart because the spectra of 2a showed two kinds of signals at δ-33(m) and −43 - −38 (m) though that of 2b showed only the signal at δ −43 - −38 (m). The former signal of 2a indicates the existence of blanched Si chain. Thus the use of stronger Lewis acid such as FeCl2 resulted in the partial cleavage of bond between Si and aromatic ring and formation of blanched 2. These results show that ZnCl2 is the best catalyst to obtain linear polysilane with an ideal chemical structure.
![]()
Table 1. Effects of catalysts on PS (2) synthesis.
![]()
Table 2. Effect of reaction temperature on the reduction of 1a.
aConditions: [Monomer 1] = 0.33 mol/l. bPurified by reprecipitation from toluene-IPA. Material yield based on 1. cDetermined by GPC based onpolystyrene standard. dNo precipitate was obtained after usual reprecipitationprocedure.
![]()
Table 3. Effect of metal chloride catalysts in preparation of 2a.
aConcentration of monomaer 1 is 1.0 mol/l. bMaterial yield based on 1. Purified by reprecipitation from toluene-IPA. cDetermined by GPC based on polystyrene standard. dUV spectra (lmax) was measured in 10% THF solution.
![]()
![]()
![]()
Figure 3. GPC charts of poly(methylphenylsilane) (2).
![]()
Figure 5. 1H-NMR and 29Si-NMR Charts of 2.
3.2. Synthesis of Various Types of Polysilanes
As shown in the Table 4, the method was effective for the synthesis of PS having at least one aromatic substituent (runs 1, 2, 4) but not effective for the synthesis of PS without aromatic substituent (run 3).
The analysis by FD-MS showed that the structure of 3 was not linear but cyclic, the mixture of 5- and 6- membered ring (Figure 6). 6 that has a linear structure with low molecular weight is known to be suitable for coating materials for PE and other alkyl polymers [28] - [30] .
3.3. Consideration of the Reaction Mechanism
The analysis of the structure of the intermediates was performed after the transformation of terminal chloro groups to ethoxy groups because of the high reactivity of chloro groups (Scheme 2).
The analyzed results on FD-MS and GPC charts were shown in Figure 7.
At the initiation stage (2 h), the results of FD-MS and GPC showed that linear oligomers were mainly formed and the high polymer was not formed. This indicated that 1 was first reduced by Mg and formed reactive species, reacting with another 1 molecule at the initiation stage. In the next stage, oligomers having Mw = ~1000 (GPC) were formed along with the formation of some high polymers. The formation of cyclic oligomers from 4-memberd to about 10-membered ring (mainly 5-memberd ring) was also observed by FD-MS charts from 4 h later. These results may show that the reaction of one oligomer with 1 or another oligomer formed PS having higher molecular weight. The intramolecular reaction of oligomers existing two terminal Cl atoms with each other resulted in the formation of cyclic oligomers (Scheme 3).
![]()
Table 4. Synthesis of a variety of polysilanesa.
aConcentration of monomer is 1.0 mol/l. bDetermined by GPC based on polystyrene standard. cMaterial yield based on the monomer. Purified by reprecipitation from toluene-IPA. dTMS shows trimethylsilylgroug.
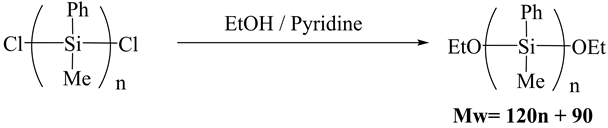
Scheme 2
![]()
![]() *In the early stage, linear oligomers were generated. **Along with polymerization, polysilanes with high molecular weight and cyclic oligomers were generated.
*In the early stage, linear oligomers were generated. **Along with polymerization, polysilanes with high molecular weight and cyclic oligomers were generated.![]()
Figure 7. Relation of reaction time and product structures (FD-MS and GPC).
Two types of mechanism may be proposed to this Si-Si bond forming reaction. Namely, one is a radical coupling reaction in which a silyl radical formed by one electron reduction of the starting chlorosilane couples with another silyl radical to give the disilane. On the other hand, in the second mechanism, two-electron reduction of the chlorosilane yields an active species equivalent to silyl anion which reacts with another chlorosilane to give the disilane. In order to have an insight into the mechanism, PS (5) obtained in the mixture of dichloromethylphenylsilane (P) and dichlorohexylmethylsilane (H) (P:H = 1:1, run 4 in Table 4) were studied in detail using FD-MS spectra (Scheme 4, Figure 8). As shown in Figure 8, the mixed coupling products (Pn - Hm) were just formed, whereas the homocoupling products (Pn) were not really found. This result seems to agree with the anion mechanism. Since homocoupling products (Hn) were not obtained shown by run 3 in Table 4, this result shows that only P can be reduced and H cannot be reduced under this reaction condition. If the reaction mechanism is a radical coupling, that is, if P is reduced to yield a radical, only Pn can be produced, and the formation of the mixed coupling products (Pn - Hm) cannot be obtained. It is indicated that the anion mechanism is more reasonable in this coupling reaction (Scheme 5).
![]()
Polysilane high polymer Cyclic oligomers
Scheme 3
![]()
Scheme 4
![]()
Figure 8. Constitution of reaction products P and H mixtures (FD-MS spectra).
Although the mechanism of the formation of PS, especially, the role of LiCl, is not always clear, one of the plausible mechanisms is as follows; The reduction of ZnCl2 with Mg [(1) in Scheme 6] forms the activated Mg (Mg*), namely, surface of Mg metal is activated by this reaction, and dichlorosilane is also activated by ZnCl2 [(2) in Scheme 6]. The reduction of activated dichlorosilane (7) with Mg* affords the corresponding silyl anion having Mg counter ion (8) [(3) in Scheme 6]. The nucleophilicity of 8 is not enough to react withother dichlorosilanes, however, the cation exchange reaction of 8 to 9 may take place in the presence of LiCl [(4) in Scheme 6]. The nucleophilicity of 9 is enough to react with other dichlorosilane molecules because of the same as the conventional Kipping method and finally form PS [(5) in Scheme 6].
3.4. Synthesis of Silane-Styrene Copolymer
Silane-styrene copolymers (10) as another type of PS can be easily formed by attacking a styrene monomer from the silyl anion when the mixture of 1 and styrene monomer was performed in the same reaction condition as PS synthesis (Scheme 7).
![]()
Scheme 5
![]()
Scheme 6
![]()
Scheme 7
![]()
Figure 9. GPC Charts of Silane-styrene Copolymers (10).
The mixture of 1 and styrene monomer was reacted in the presence of Mg powder, anhydrous LiCl, ZnCl2 in dried THF similar to PS synthesis. A white powder was obtained after purifying by reprecipitation from IPA (10a).
The GPC chart of 10a suggests that it was polymeric materials as shown in Figure 9. The 29Si-NMR spectrum of this polymer is shown in Figure 10. Three Si peaks were observed. If this polymer is a mixture of polysilane and polystyrene, the Si peak would be one peak because it becomes the same as polysilane peak (Figure 10(1)) δ ?44 - ?36). On the other hand, if it is a silane-styrene copolymer, it would be found three peaks as showed below.
1) Both sides of Si are styrene units.
2) One side of Si is Si and another side is a styrene unit.
3) Both sides of Si are Si units.
Accordingly, it can be conjectured that this provided polymer is objective silane-styrene copolymers. The ratio of three peaks shows almost the same value and hence 10a indicates almost the random copolymers.
As a next experiment, the formation of block copolymers was performed. Namely, 1 was first reacted with Mg for several hours to form silane-oligomers and then styrene monomers were added later. The results of GPC and 29Si-NMR charts obtained polymers were shown in Figure 9 and Figure 10 respectively. In case of these polymers, 29Si-NMR also showed the existence of three different types of Si peaks. However, as the adding time of styrene becomes later, peak intensity of a) and b) decreased to that of c). These results indicate 10b and 10c were undoubtedly block copolymers with 1 and styrene monomers.
4. Conclusions
The synthetic method of polysilane using Mg and Lewis acids was highly practical since it was carried out under mild reaction conditions. It was also applied for the synthesis of various types of PS having a linear structure, a cyclic structure, liquid property that is suitable for coating materials and silane-styrene copolymers as another type of PS. The reaction mechanism was also elucidated. The analysis of the reaction intermediates was performed, and the anion mechanism but not the radical one would be reasonable in this Si-Si bond forming reaction.
![]()
Figure 10. 29Si-NMR Charts of Silane-styrene Copolymers (10).
Acknowledgements
Work of preparation of polysilane was supported by Kindai University in Japan. The authors gratefully acknowledge to Professor Shigenori Kashimura, Faculty of Engineering, Kindai University, in Japan.
NOTES
*Corresponding author.