A Comparative Study on the Structural and Vibrational Properties of Two Potential Antimicrobial and Anticancer Cyanopyridine Derivatives ()
1. Introduction
As part of our investigations on compounds of great pharmacological interest [1] , in this work we have studied from a theoretical point of view the structures and vibrational properties of the 2-hydroxy-4,6-dimethylpyridine- 3-carbonitrile and 2-chloro-4,6-dimethylpyridine-3-carbonitrile compounds. These cyanopyridine derivatives play a very important role in the synthesis chemistry organic and, in medicine and pharmacology because these compounds present potential anticancer, antiarrhythmic, anticonvulsant, antiparkinsonian and antimicrobial activities [2] -[8] . Recently, Waly et al. have reported the synthesis and antitumor evaluation of some new fused and binary pyridines together with a modified synthetic method for the synthesis of 2-chloro-4,6-dimethyl-nico- tinonitrile [2] . In other studies, the synthesis and antiarrhythmic activities of some synthesized pyridine derivatives fused with thiophene ring were also reported while new pyridine derivatives were synthesized and evaluated as analgesic, anticonvulsant and anti-parkinsonian agents [4] . Besides, derivatives as the thieno [2,3-d] pyrimidines can be used to eliminate fungi in plants [9] and to inhibit the growth of cancer cells [10] while some thiopyridines and carbonitrile compounds are also used as insecticides [11] -[13] . All these studies clearly show that the modifications in the pyridine structure by incorporation of different groups and/or rings produce different biochemical behavior and biological activity that are evidenced by different structure-activity relationships (SAR). Hence, the studies of the structural properties of these derivatives are important to predict their reactivities and behavior in different media and systems which are present, especially when these compounds are used as pharmacological drugs for the health human. Furthermore, the knowing of its structures is also very important because it permits their quickly identifications by means of vibrational spectroscopy. So far, the crystal and molecular structures of both compounds were not determined and only the structure of 2-anilino-4,6-dimethylpyri- dine-3-carbonitrile, an intermediate in the synthesis of 5-aminobenzo [b] [1] [8] naphthyridines was published by Mefetah et al. [14] . On the other hand, the crystal structure of 4-cyanopyridine was determined by Laing et al. [15] from three-dimensional single crystal X-ray data collected by standard film techniques while the structure of the 4-cyanopyridinium dihydrogen phosphate-isonicotinonitrile-phosphoric acid compound was refined by Wang [16] who has determined that this compound has a asymmetric unit because it contains one 4-cyanopyri- dinium cation, one 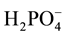 anion, one independent isonicotinonitrile molecule and one independent H3PO4 molecule. Also, from long time the cyano group structure in 3-cyanopyridinium tetrachloroferrate (III)-3-cya- nopyridine was determined by Daran et al. [17] by X-ray diffraction method. In the present work, we presented the structural and vibrational studies of the 2-hydroxy-4,6-dimethylpyridine-3-carbonitrile and 2-chloro-4,6- dimethylpyridine-3-carbonitrile compounds in order to, 1) determine their theoretical and more stable structures in gas and aqueous solution phases, 2) perform complete assignments of the corresponding infrared and Raman spectra, 3) evaluate their structural properties in gas and aqueous solution phases and, 4) predict the behavior of both molecules in gas and aqueous solution phases by using diverse descriptors [18] [19] . For those purposes, first, both structures were optimized in gas and aqueous solution phases using the hybrid B3LYP/6-31G* method and the solvent effects in aqueous solution were studied by using the self-consistent reaction field (SCRF) calculations with the IEFPCM model [20] . Then, the corresponding frequencies were calculated at the same level of theory in order to perform the complete assignments of all the bands observed in the vibrational spectra combining the natural internal coordinates with the scaled mechanical force field (SQMFF) procedure [21] . Additionally, molecular electrostatic potentials (MEP), highest occupied molecular orbital-lowest unoccupied molecular orbital (HOMO-LUMO) [22] , natural bond orbital (NBO) [23] [24] and atoms in the molecules (AIM) [25] [26] calculations were performed in order to evaluate the structural properties such as, electrostatic potentials, atomic charges, bond orders, stabilization energies, topological properties and solvation energies in both media, and to predict the properties of the two compounds in both media. Here, the structural and vibrational properties of both compounds were compared and analyzed.
anion, one independent isonicotinonitrile molecule and one independent H3PO4 molecule. Also, from long time the cyano group structure in 3-cyanopyridinium tetrachloroferrate (III)-3-cya- nopyridine was determined by Daran et al. [17] by X-ray diffraction method. In the present work, we presented the structural and vibrational studies of the 2-hydroxy-4,6-dimethylpyridine-3-carbonitrile and 2-chloro-4,6- dimethylpyridine-3-carbonitrile compounds in order to, 1) determine their theoretical and more stable structures in gas and aqueous solution phases, 2) perform complete assignments of the corresponding infrared and Raman spectra, 3) evaluate their structural properties in gas and aqueous solution phases and, 4) predict the behavior of both molecules in gas and aqueous solution phases by using diverse descriptors [18] [19] . For those purposes, first, both structures were optimized in gas and aqueous solution phases using the hybrid B3LYP/6-31G* method and the solvent effects in aqueous solution were studied by using the self-consistent reaction field (SCRF) calculations with the IEFPCM model [20] . Then, the corresponding frequencies were calculated at the same level of theory in order to perform the complete assignments of all the bands observed in the vibrational spectra combining the natural internal coordinates with the scaled mechanical force field (SQMFF) procedure [21] . Additionally, molecular electrostatic potentials (MEP), highest occupied molecular orbital-lowest unoccupied molecular orbital (HOMO-LUMO) [22] , natural bond orbital (NBO) [23] [24] and atoms in the molecules (AIM) [25] [26] calculations were performed in order to evaluate the structural properties such as, electrostatic potentials, atomic charges, bond orders, stabilization energies, topological properties and solvation energies in both media, and to predict the properties of the two compounds in both media. Here, the structural and vibrational properties of both compounds were compared and analyzed.
2. Computational details
Initially, the structures of the 2-hydroxy-4,6-dimethylpyridine-3-carbonitrile (2-OH) and 2-chloro-4,6-dime- thylpyridine-3-carbonitrile (2-Cl) derivatives were modeled with the GaussView program [27] and optimized by using the hybrid B3LYP/6-31G* method [28] [29] employing the Gaussian 09 program [30] . For each (2-OH) and (2-Cl) derivative only a stable conformation of C1 symmetry was optimized which can be seen in Figure 1 together with the labelling of the atoms. In aqueous solution, the solvent effects were simulated employing the SCRF method by using the IEFPCM model [20] with a value of 78.39 to simulate the aqueous medium. This way, the geometrical parameters and frequencies for those derivatives were calculated at the same level of theory in order to confirm the nature of the stationary points. Also, the solvation energies were calculated from the IEFPCM calculations with radii and non-electrostatic terms by using the SMD solvation model [31] , as re-
![]()
Figure 1. Theoretical structures and atoms numbering for the: (a) 2-Hydroxy-4,6-dimethylpyridine-3-carbonitrile and; (b) 2- Chloro-4,6-dimethylpyridine-3-carbonitrile derivatives.
ported for other molecules in aqueous medium [32] -[34] .
For both derivatives, the variations of molecular volumes expressed as a difference between the volumes in aqueous solution in relation to the volume in gas phase were calculated employing the Moldraw program [35] . In order to know the exact nature of the interactions present in both derivatives the atomic charges and those derived from Merz-Kollman were considered in this study [36] together with the corresponding MEP. In addition, the bond orders were calculated at the same theory level from NBO calculations by using the NBO 3.1 program [24] , as implemented in the Gaussian 09 package [30] . The different interactions predicted for both derivatives in the two studied media were analyzed by means of calculations of the electronic charge density topological with the
AIM
200 program package [26] . The harmonic force fields for both derivatives were evaluated at the same level of theory by using the Molvib program [37] and following the SQMFF procedure [21] . To perform the complete assignments, only the potential energy distribution components (PEDs) ³10% were considered from the resulting SQM.
3. Results and discussion
3.1. Geometry
Table 1 show the calculated geometrical parameters for the two studied cyanopyridine derivatives in gas and aqueous solution phases by using the B3LYP/6-31G* level of theory. The theoretical values were compared with the experimental ones determined by Mefetah et al. for 2-anilino-4,6-dimethylpyridine-3-carbonitrile by using X-ray diffraction method by means of the root mean square deviation (RMSD) [14] . The results show that for both molecules the calculated bond length and angles values are in agreement with the experimental ones with RSMD values for bond length between 0.004 and 0.005 Å while the difference for the bond angles are between 0.7˚ and 0.8˚. Comparing the calculated parameters for the two derivatives with those experimental obtained for 4-cyanopyridine [15] , we observed that the experimental CºN distance in it derivative is 1.137 Å while in the 4-cyanopyridinium dihydrogen phosphate-isonicotinonitrile-phosphoric acid derivative [16] is 1.142 Å and in 3-cyanopyridinium tetraehloroferrate(lll)-3-cyanopyridine is of 1.102 Å [17] .
The results clearly show that this bond is strongly dependent of the groups linked to the pyridine ring and of the position of the C-CºN group. On the other hand, the C-C bond linked to the CºN bond in 4-cyanopyridine is
![]()
Table 1. Comparison of calculated geometrical parameters for the two studied cyanopyridine derivatives.
aThis work; bFrom Ref [38] .
1.439 Å [15] , in 4-cyanopyridinium dihydrogen phosphate-isonicotinonitrile-phosphoric acid [16] is 1.453 Å and in 3-cyanopyridinium tetraehloroferrate (III)-3-cyanopyridine [17] is between 1.452 and 1.480 Å, being the calculated values for (2-OH) and (2-Cl) between 1.428 and 1.422 Å. Thus, these values indicating that this bond is less depending of the position of the C-CºN group and of the groups linked to the pyridine ring. Also, in both derivatives we observed that the C1-N7 distances are slightly different from the C5-N7 distances indicating that both pyridinic bonds are dependent of the groups linked to the C1 or C5 atoms belonging to the C-N bonds, these are, in the (2-OH) derivative, C1-O18 and C5-CH3 while in the (2-Cl) derivative are C1-Cl18 and C5-CH3. Experimentally, the C-N distance in the symmetric molecule of 4-cyanopyridine [15] is 1.331 Å, in 4-cyanopyri- dinium dihydrogen phosphate-isonicotinonitrile-phosphoric acid [16] are 1.339 and 1.337 Å and, in 3-cyanopy- ridinium tetraehloroferrate(III)-3-cyanopyridine [17] those distances are between 1.301 and 1.351 Å. The differences observed in the latter molecule are justified because two molecules are linked in asymmetric form by the N atoms of both pyridine rings. Also, in the (2-OH) and (2-Cl) derivatives (Table 1) the two C-N distances are different because both molecules are asymmetrics. In aqueous solution, we observed that in both derivatives some distances slightly change with the hydration, being more evident in the C1-O18 and C1-Cl18 distances because both are sites of H bonds formation. In the (2-OH) derivative, the C-OH site is an electrophilic region while in the (2-Cl) derivative the C-Cl site is a nucleophilic region. On the other hand, Table S1 show a comparison of the total energies and the corresponding dipole moment values for both derivatives in the two media by using the B3LYP/6-31G* method. Note that in both media the dipole moments for the (2-Cl) derivative are higher than the other ones, as expected because the Cl atom is a voluminous atom. For this reasons, the calculated molecular volumes for the (2-Cl) derivative in both media by using the Moldraw program [35] and the B3LYP/6-31G* method are higher than the other ones, as observed in Table 2. In the (2-Cl) derivative, clearly there is a volume contraction with the hydration while in the (2-OH) derivative is observed a volume expansion, in agreement with the solvation energies values, as can be seen in Table 2.
3.2. Solvation energies
The uncorrected solvation energies (DGu), calculated as relative energies (DE) and defined as the difference between the total energies in aqueous solutions and the values in gas phase for the (2-OH) and (2-Cl) derivatives using the 6-31G* basis set, are presented in Table 2. Here, the uncorrected (DGu) and corrected (DGc) solvation energies together with the total non electrostatic terms (DGne) due to the cavitation, dispersion and repulsion energies were calculated by using the PCM/SMD model [31] , in form similar as reported for other molecules in aqueous solution [32] -[34] . Note that for the (2-OH) derivative a higher DGc value is obtained, probably due to its higher variation of volume in solution (Table 2), for this reason, a higher hydration is expected for this derivative in aqueous solution.
3.3. Molecular electrostatic potential, atomic charges and bond orders
The molecular electrostatic potential values for both derivatives calculated in the two media by using the B3LYP/6-31G* method are given in Table S2. In (2-OH), the most negatives molecular electrostatic potential values are observed on the N7, N17 and O18 atoms and the less negative on the H19 atom belonging to the OH group while in (2-Cl), the most negatives values are observed on the N7, N17 and Cl18 atoms and the less negative on the H atoms. Note that in both molecules the N17 atom have higher values than the N7 ones, for these rea- sons, strong red colorations are observed on the surfaces mapped of both derivatives, as observed in Figure S1, indicating in both derivatives those regions reacting with potential biological electrophiles. These colorations indicate probably capability of hydrogen bond formation with share of nitrogen atoms of the CºN or, in aqueous solution through hydrogen atom of -OH moiety. On the contrary, on the H19 atom a strong blue coloration is observed on the surfaces mapped of (2-OH) being this region reacting with potential biological nucleophiles. Another important result is the lower molecular electrostatic potential values observed on the H atoms of both
![]()
Table 2. Calculated volume and solvation energies (DG) for the two studied cyanopyridine derivatives.
DGc = DGuncorrected# − DGTotalnon electrostatic; aThis work.
CH3 groups in the chlorinated derivative in the two media in relation to the other one. These results justify the higher blue coloration on the surface mapped of those groups in the chlorinated derivative, as observed in Figure S1. In the (2-OH) derivative is expected a high reactivity due to the higher proximity between the N17 and O18 atoms (higher repulsion), whose distances in gas and aqueous solution phase are respectively of 3.559 and 3.496 Å while in the chlorinated derivative the N17-Cl18 distances in gas and aqueous solution phases are respectively of 3.743 and 3.698 Å. Note that in aqueous solution increase the reactivities of both derivatives due to the shortening in the distances between both involved atoms. The atomic charges derived from the ESPs (MK) [36] and the NPA charges by using the B3LYP/6-31G* method were calculated for both derivatives in the two media, as can be seen in Table S3. The two charges show that despite the only difference between both derivatives are the presence of the OH group and the Cl atom the values are strongly dependent of the groups linked to the pyridine ring and of the position of the C-CºN group, as was also observed in the Section 3.1. These charges analysis also show that the (2-OH) derivative is less stable than the other one due to the higher charge values on the N17 and O18 atoms. Moreover, the different charge values observed on the N7, N17, O18 and Cl18 atoms suggest for the two derivatives a different hydration in solution. The bond orders expressed by Wiberg’s indexes for the two derivatives in both media are observed in Table S4. Note that the change of the OH group in (2-OH) by the Cl atom in (2-Cl) increase the bond order values for the N7, N17 and all the C atoms of the pyridine ring while decrease the bond order values of the H and C atoms belonging to the two CH3 groups, thus, the pyridine ring has in gas phase a higher stability in the chlorinated derivative than the other one. Note that the bond order values in both derivatives practically not change in aqueous solution and only a little decreasing is observed in the bond order corresponding to the Cl atom with the hydration. This fact, in (2-Cl) is probably related with the contraction volume observed in aqueous solution.
3.4. NBO Analysis
The stability of both derivatives were studied by means of second order perturbation energies E(2) (donor à acceptor) whose values are given in Table S5. Here, we considered only those contributions higher than 20 kJ/mol. These results for the (2-OH) and (2-Cl) derivatives show three different contributions to the total stabilization energies, which are, the DETp®p*, DETLP®s* and DETs*®s* charges transfer, being the two latter interactions higher in the (2-OH) derivative than the other one while, the π-π interactions in the pyridine ring are higher in the chlorinated derivative. These results show that the calculated total stabilization energy favours to the (2-OH) derivative revealing thus a higher stability for this derivative in gas and aqueous solution phases. Here, it is very important to note that in the (2-OH) derivative the significant increase in the delocalization values in aqueous solution is related with the increase of the C-C and C-N double bonds of the pyridine ring as consequence of the hydration. This way, this analysis shows clearly that the DETπ-π interactions in the pyridine ring are higher in the chlorinated derivative and in both media while, in general, the DETotal are higher in the (2-OH) derivative than the other one.
3.5. AIM study
Both derivatives were also studied employing the AIM analysis [25] [26] . For the (2-OH) and (2-Cl) derivatives, the charge electron densities, (r) and the Laplacian of the electron densities, Ñ2r(r) were calculated for the ring critical points (RCPs) belonging to the pyridine rings and the values can be seen in Table S6. The results show higher values in the topological properties for the chlorinated derivative and, also a slightly difference for this derivative with the hydration. Thus, the analysis clearly shows a higher stability for the pyridine ring of the chlorinated derivative in both media, contrarily to that result obtained by NBO analysis.
3.6. Descriptors for both derivatives
Many cyanopyridine derivatives have potentials antimicrobial and anticancer activities, for this reason, and to determine the exact nature of the interactions with electrophones and/or nucleophiles and, also to predict the behavior of both derivatives in gas and aqueous solution phases are very important the calculations of some descriptors. If both derivatives are used for the drugs design, the knowledge of these descriptors helps to understand the structural, dynamical, and functional properties of each derivative in both media. Thus, for both derivatives, the HOMO and LUMO orbitals, energy band gap, chemical potential (μ), electro negativity (χ), global hardness (η), global softness (S) and global electrophilicity index (ω) descriptors [18] [19] were calculated. These descriptors calculated in both phases at B3LYP/6-31G* level of theory can be seen in Table S7. Comparing first the energy band gaps between both derivatives we observed that the chlorinated derivative has the highest values in both media indicating a higher stability for this molecule than the other one. Note that the OH group in the pyridine ring generate the reduction of the HOMO-LUMO gap indicating that in the (2-OH) derivative that group deactivating the ring and diminishing its potency when it is used as a drug. On the contrary, the presences of a Cl atom in the pyridine ring in (2-Cl) compared with (2-OH), active that ring and increase its potency. Comparing the calculated chemical hardness η, chemical potential m and global electrophilicity index ω values for (2-OH) with those obtained for (2-Cl) in both phases, we observed that (2-Cl) is more stable in both media (larger η) and has better capability to accept electrons (bigger electrophilicity index) than (2-OH) but, on the contrary, (2-OH) is better electrons donor than (2-Cl). Here, the lowest molecular electrostatic potentials observed on the H atoms of both CH3 groups for the chlorinated derivative support the better capability to accept electrons while, the highest molecular electrostatic potentials observed on the N7 and N17 atoms of (2-OH) suggest that it derivative is better electrons donor than (2-Cl).
3.7. NMR analysis
Experimental and calculated chemical shifts with the GIAO method using 6-311++G** basis set for the 1H and 13C nuclei of both derivatives are compared in Table S8 and Table S9 respectively. In general, the calculated shifts for the 13C nuclei are lower than the corresponding experimental values. Note that the calculated chemical shifts for the H nuclei of (2-OH) show a significant variation (0.81 ppm) than the (2-Cl) derivative (0.22 ppm), in relation to the corresponding experimental values [2] . Probably, the theoretical calculations do not correctly predict the hydrogen chemical shift of the H19 nucleus belonging to the OH group of (2-OH), as observed in Table S8, because that group is involved in intermolecular H-bonds. Thus, the higher hydration of this derivative is supported by the higher solvation energy in aqueous solution. Table S9 show that the calculated 13C chemical shifts for both derivatives are slightly different between them, as expected due to the different (OH and Cl groups) present in each structure.
4. Vibrational analysis
The recorded infrared spectra for both derivatives in solid phase compared with the corresponding theoretical in gas and aqueous solution phases can be seen respectively in Figure 2 and Figure 3. Both infrared spectra in solid phase were taken from Refs [38] [39] . The predicted Raman spectra calculated using B3LYP/6-31G* method for (2-OH) and (2-Cl) can be seen in Figure 4. The (2-OH) and (2-Cl) derivatives have respectively 51 and 48 normal vibration modes, all active in the infrared and Raman spectra. The experimental and calculated wavenumbers for both derivatives together with the corresponding assignments are shown in Table 3. The vibrational assignments of the experimental bands to the normal vibration modes for both derivatives are based on the comparisons with related molecules [40] -[42] and with the results of the calculations performed here. In this work, we presented only the calculations for both derivatives in gas phase because the experimental spectra were not registered. The comparison between the theoretical infrared spectra for (2-OH) and (2-Cl) using B3LYP functional and 6-31G* basis in gas (black color) and aqueous solution phases (red color) can be seen in Figure S2. Note that in both spectra in aqueous solution are observed a shifting and intensification of the bands specially those assigned to the CºN, C-N, O-H stretchings and in the 1500 - 500 cm−1 region attributed to the H bonds, as observed in Figure S2. The SQM force fields for both derivatives can be obtained at request. The discussion of assignments of the most important groups for both derivatives is presented as follows.
4.1. Assignments for 2-hydroxy-4,6-dimethylpyridine-3-carbonitrile
OH modes. The broad and intense band in the IR spectrum of (2-OH) centred at 3350 cm−1 and predicted in aqueous solution at 3523 cm−1, is assigned to the O-H stretching as observed in compounds containing this group [34] [41] -[44] . The OH in plane deformation mode is assigned to the strong band at 1225 cm−1 while the corresponding out-of-plane deformation mode is associated to the band at 495 cm−1.
CH3 modes. The IR bands between at 2980 and 2850 cm−1 are assigned to the CH3 antisymmetric and sym- metric stretching modes while the bands between 1465 and 1360 cm−1 are clearly assigned to the ant symmetric and symmetric CH3 deformation modes. The four expected rocking modes are assigned to the shoulder and
![]()
Figure 2. Comparison between the experimental Infrared spectra of: (a) 2- hydroxy-4,6-dimethylpyridine-3-carbonitrile from Ref [38] ith the corres- ponding theoretical in (b) gas phase and, (c) in aqueous solution at B3LYP/6- 31G* level of theory.
![]()
Figure 3. Comparison between the experimental Infrared spectra of: (a) 2- chloro-4,6-dimethylpyridine-3-carbonitrile from Ref [39] with the corres- ponding theoretical in (b) gas phase and, (c) in aqueous solution at B3LYP/6- 31G* level of theory.
bands observed between 1095 and 1025 cm−1. The twisting modes were not assigned because are predicted at 66 and 41 cm−1.
C-CºN modes. Here, the IR band at 2220 cm−1 is assigned to the CºN stretching mode while the strong band at 720 cm−1 is assigned to the C2-C16 stretching mode, as observed in Table 3. The bending and out-of-plane deformation modes corresponding to the C2-C16 group are predicted at 236 and 138 cm−1 and, for this reason, they were not assigned.
Skeletal modes. The C-N stretching modes corresponding to the pyridine ring are predicted by the calcula-
![]()
Figure 4. Comparison between the theoretical Raman spectra of the 2-hy- droxy-4,6-dimethylpyridine-3-carbonitrile and 2-chloro-4,6-dimethylpyridine- 3-carbonitrile derivatives at B3LYP/6-31G* level of theory.
tions in different regions. Hence, the IR bands at 1480 and 1140 cm−1 are associated respectively with those two C5-N7 and N7-C1 stretching modes while the strong band at 1375 cm−1, is associated with the C-O stretching mode. Here, it is necessary to note that in aqueous solution there is a very important shifting in the wave numbers corresponding to those stretching modes, thus, the N7-C1 and C5-N7 stretching modes are predicted respectively at 1486 and 963 cm−1, as observed in Table 3. This fact is related with the higher hydration of the (2-OH) derivative and with the increasing in the corresponding force constant values with the hydration as consequence of the increasing in the N7-C1 double bond character, as we will see later. Finally, the observed torsion and deformation modes of the pyridine ring are assigned as predicted by calculations and taking into account the assignments for similar molecules [45] -[49] . In this work, those modes are identified and assigned, as observed in Table 3.
4.2. Assignments for 2-chloro-4,6-dimethylpyridine-3-carbonitrile
CH3 modes. As in the (2-OH) derivative, the CH3 ant symmetric and symmetric stretching modes are assigned between 3010 and 2975 cm−1 while the ant symmetric and symmetric CH3 deformation modes bands are clearly assigned, as predicted by calculations, to the strong band at 1440 cm−1. The four expected rocking modes are assigned to the bands between 1040 and 1000 cm−1. In this derivative, the twisting modes were not assigned because both are predicted at 77 and 57 cm−1. It is important to note that in this derivative the presence of the Cl atom in the structure shift the bands toward lower wave numbers, as observed in Table 3.
C-CºN modes. Here, the CºN stretching mode is assigned to the IR band at 2225 cm−1 while the weak band at 700 cm−1 is assigned to the C2-C16 stretching mode, as predicted by calculations.
In this derivative, with the hydration only is observed a shifting in the wave numbers related to the C5-N7 stretching mode, as indicated in Table 3. In (2-Cl), the C2-C16 bending mode is assigned to the weak band at 615 cm−1 while the corresponding out-of-plane deformation mode is predicted at 241 cm−1 and, for this, it mode is not assigned.
Skeletal modes. In this derivative, the C-N stretching modes corresponding to the pyridine ring are predicted by the calculations in the same regions, thus, both modes were assigned to the band and shoulder respectively at 1260 and 1250 cm−1. The C-Cl stretching mode is predicted by calculations at 448 cm−1 and assigned at 440 cm−1. The bending and out-of-plane deformation modes corresponding to the C1-Cl18 group are predicted at 230 and 160 cm−1, hence, these modes were not assigned. Finally, in accordance with similar molecules [41] -[49] the torsion and deformation modes corresponding to the pyridine ring are assigned as predicted by calculations, as observed in Table 3.
![]()
![]()
Table 3. Observed and calculated wavenumbers (cm−1) and assignments for the two studied cyanopyridine derivatives in gas phase and aqueous solution.
n, stretching; d, scissoring; wag and g, wagging or out of plane deformation; r, rocking; t, torsion, twist, twisting; a, antisymmetric; s, symmetric; R, ring; aThis work, bFrom Ref [38] , cFrom Ref [39] , dFrom scaled quantum mechanics force field B3LYP/6-31G*, eFrom scaled quantum mechanics force field PCM/B3LYP/6-31G*.
5. Force field
The force constants were calculated from the corresponding scaled force fields by using the Molvib program [37] , as was described in Section 2. A comparison of the principal force constants for both derivatives in the two studied media are given in Table 4. In this study, the higher modifications are observed in the f(nC-N), f(nC-C) and f(nC-O) force constants related to the (2-OH) derivative in aqueous solution because the values increasing notably with the hydration. In the (2-Cl) derivative only a slight increasing in some values and decreasing in other were observed with the hydration. Note that those higher values observed in the (2-OH) derivative in aqueous solution can not be attributed to the geometrical parameters because the C5-N7 and N7-C1 distances not change with the hydration, as observed in Table 1 while, on the contrary, the C1-O8 distance is enlarged in solution. Thus, those values can be justified in part by the NBO results because there is a strong delocalization on the pyridine ring in the (2-OH) derivative that increasing its stability in aqueous solution due to the increasing in the double bonds character. The shifting of the bands attributed to the C5-N7, N7-C1 and C1-O8 stretching modes with the hydration also support the increasing of the corresponding force constants values.
6. Conclusion
The theoretical molecular structures of the 2-hydroxy-4,6-dimethylpyridine-3-carbonitrile and 2-chloro-4,6-di- methylpyridine-3-carbonitrile derivatives were determined in gas phase and in aqueous solution by using the B3LYP/6-31G* method employing the IEFPCM model. The complete assignments of the vibrational modes for both derivatives and the corresponding SQM force fields were obtained by using the B3LYP/6-31G* method. The predicted Raman spectra for the 2-hydroxy-4,6-dimethylpyridine-3-carbonitrile and 2-chloro-4,6-dime- thylpyridine-3-carbonitrile derivatives have been reported by using the B3LYP/6-31G* method. Differences in
![]()
Table 4. Scaled force constants for the two studied cyanopyridine derivatives in gas and aqueous solution phases.
n, stretching; δ angle deformation. Units in mdyn Å−1 for stretching and mdyn Å rad −2 for angle deformations; aThis work.
the studied properties for both derivatives in both media were justified by the molecular electrostatic potentials, atomic charges, bond orders, solvation energies, dipole moments, deslocalization energies and AIM analysis. A higher stability in aqueous solution for the 2-hydroxy-4,6-dimethylpyridine-3-carbonitrile derivative was found, which is supported in part by the NBO analysis, by a higher hydration of this derivative in solution due to its higher solvation energy and, by the higher force constant values. The analysis of the descriptors suggests that the OH group in the 2-hydroxy-4,6-dimethylpyridine-3-carbonitrile derivative reduces the HOMO-LUMO gap deactivating the ring while the calculated chemical hardness, chemical potential and global electrophilicity index values suggest a higher stability for the 2-chloro-4,6-dimethylpyridine-3-carbonitrile derivative and a better capability to accept electrons, as suggested by the AIM analysis. Here, the differences observed between the NBO and AIM results are probably due to that in the total energy only were considered those contributions with values higher than 20 kJ/mol. 1H-NMR spectra observed for both derivatives were successfully compared with the calculated chemical shifts at the B3LYP/6-311++G** level of theory. The high value observed in the hydrogen chemical shift corresponding to the H atom of the OH group of 2-hydroxy-4,6-dimethylpyridine-3-carbonitrile, in relation to the calculated value, confirms the presence of the hydrogen bonds in solution for this derivative.
Acknowledgements
This work was founded with grants from CIUNT (Consejo de Investigaciones, Universidad Nacional de Tucumán). The authors thank Prof. Tom Sundius for his permission to use MOLVIB.
Supporting Information
![]()
Table S1. Total (E) and relative (DE) energies and dipole moment (m) for the two studied cyanopyridine derivatives.
![]()
Table S2. Molecular electrostatic potential (in a.u.) for the two studied cyanopyridine derivatives.
![]()
![]()
Table S3. Atomic charges for the two studied cyanopyridine derivatives.
![]()
Table S4. Wiberg indexes for the two studied cyanopyridine derivatives.
![]()
![]()
Table S5. Main delocalization energy (in kJ/mol) for the two studied cyanopyridine derivatives.
![]()
Table S6. Analysis of the ring critical points (RCP) for the two studied cyanopyridine derivatives.
![]()
![]()
Table S7. Calculated HOMO and LUMO orbitals, energy band gap, chemical potential (μ), electronegativity (χ), global hardness (η), global softness (S) and global electrophilicity index (ω) for the two studied cyanopyridine derivatives.
![]()
![]()
Table S8. Observed and calculated hydrogen chemical shifts (d, in ppm).
aGIAO/B3LYP/6-311++G** Ref. to TMS; bExperimental dissolved in CDCl3 [2] . TMS as reference.
![]()
Table S9. Observed and calculated hydrogen chemical shifts (d, in ppm).
aGIAO/B3LYP/6-311++G** Ref. to TMS.
![]()
Figure S1. Calculated electrostatic potential surfaces on the molecular sur- faces of 2-OH and 2-Cl derivatives. Color ranges, in au: from red −0.05 to blue +0.05. B3LYP functional and 6-31G* basis set. Isodensity value of 0.005.
![]()
Figure S2. Comparison between the theoretical infrared spectra in gas phase (black color) and in aqueous solution (red color) at B3LYP/6-31G* level of theory for 2-hydroxy-4,6-dimethylpyridine-3-carbonitrile (upper) and 2-chloro- 4,6-dimethylpyridine-3-carbonitrile (bottom).