Theoretical Study of Benzothiazole and Its Derivatives: Molecular Structure, Spectroscopic Properties, NBO, MEP and TD-DFT Analyses ()
1. Introduction
Compounds from the Benzothiazole family are synthesized in large quantities, due to their widespread spread use in industrial and domestic fields. These compounds are also used in many varieties of fields, including agriculture, the detergents industry, the papers industry, and the automotive and pharmaceutical industries [1] [2] . Their release into the environment causes a pollution problem [3] [4] . They are classified among emerging environmental pollutants with a high production volume [5] [6] . Their presence in domestic wastewater has been demonstrated [7] [8] . Toxicity studies have shown that these pollutants present a risk to human being health.
In particular, exposure to high concentrations of benzothiazole can have harmful effects on the liver and kidneys and cause respiratory irritation [9] [10] . Food, drink and pharmaceutical products are the most common routes of human exposure to these pollutants. Tobacco smoke isn’t the only gateway for harmful chemicals to enter our bodies. Inhalation, as you rightly pointed out, presents a broader concern, with several other routes posing serious health risks [9] .
Here’s a reformulated text without plagiarism about the link between their presence in the atmosphere and tyre wear, rubber degradation [11] [12] and the use of biocides and anti-freeze against corrosion [8] . Benzothiazole and its derivatives are therefore found in the air in particulate or gaseous form [9] .
In order to reduce the level of pollution and solve the health problems due to this type of noxious substance, researchers have been interested in the biodegradation and photodegradation of these compounds [7] [13] [14] . To truly understand how these pollutants vanish naturally, delving into the realm of computational modeling and their inherent reactivity becomes crucial. Such theoretical studies act as a powerful microscope, peering beyond the limitations of lab experiments and revealing the intricate secrets of biodegradation. Density Functional Theory (DFT) is one of the most popular computational methods in the field of quantum chemistry the most powerful tools available for elucidating adsorption processes in adsorption materials at the molecular level and for stability and reactivity studying. The reactivity and stability of benzothiazole and some of its derivatives in the gaseous and aqueous phases are studied.
Sequence to highlight detailed information on the reactivity of benzothiazole and its derivatives, the MEP surface map was determined. Other parameters such as descriptors of the chemical reactivity, chemical potential μ, electronegativity χ, hardness ƞ, electrophile number ω, we have undertaken a rigorous analysis, calculating various chemical reactivity descriptors to scrutinize them in detail. This examination promises to deepen our understanding of these molecules’ behavior and potential interactions. The final stage of our study focused on the stability of benzothiazole and its derivatives, both in the ethereal world of gases and in the dynamic world of aqueous environments. Using a powerful combination of thermodynamic and charge transfer parameters, we meticulously assessed the forces governing their stability. In particular, we have integrated the subtle influences of hyperconjugative interactions and electronic transitions through the lens of NBO calculations, providing a more nuanced understanding of the stability landscape.
2. Materials and Methods
Theoretical quantum chemical calculations we employed Gaussian 09, a computational chemistry software, to perform the density functional theory (DFT) simulations [15] and visualized through its software called gauss view 6.0 [16] . We optimized molecular geometries using density functional theory (DFT) with the 6-31+G (d, p) basis set [17] and employing the Becke exchange functional (B) [18] which combines the following elements Lee, Yang and Parr (LYP) [19] . Specifically, we calculated the dipole moment, HOMO energy (EHOMO), LUMO energy (ELUMO), HOMO-LUMO gap, electrostatic potential surface, and key global reactivity indices like ionization potential and electrophilicity. These parameters were calculated by using the hybrid density functional theory (DFT) method with the B3LYP functional, calculations were performed on the optimized geometries of the molecule in both gas and aqueous phases, simulated at standard temperature (298.15 K) and pressure (1 atm). To account for the solvation effects of water, we employed the CPCM solvation model at the dielectric constant of 78.39, implicitly incorporating the solvent without explicit water molecules [20] . Our theoretical approach involved B3LYP/6-31+G (d, p) calculations on the ground state geometry, ensuring accurate evaluation of thermodynamic parameters for further analysis. NBO analysis was conducted on the optimized structures obtained at the same level of theory (B3LYP/6-31+G (d, p)) to generate wave function files, enabling detailed insights into the electronic bonding and interactions within the molecule. Excitation energies, wavelengths, oscillation strengths, total energies, etc. were also obtained using TD-DFT (time- dependent density functional theory) Additionally, time-dependent density functional theory (TD-DFT) calculations, as described in references [21] [22] , were employed to determine the electronic excitation energies in both gas and aqueous phases. This allowed us to estimate the stabilization energy (E2) associated with electron delocalization between each donor and acceptor NBO pair (i and j):
This expression represents the second-order interaction energy (E2) between occupied orbitals “i” and “j”, calculated using the diagonal elements (qi) and off-diagonal elements (Fi;j) of the Natural Bond Orbital (NBO) Fock matrix. Analyzing the molecular electrostatic potential (MEP) maps, visualized as negative (red) and positive (blue) regions, allowed us to identify potential electrophilic and nucleophilic attack sites on the different compounds, respectively. To assess the influence of lipophilicity, we determined the partition coefficient (P), expressed as the logarithm of the concentration ratio of the solute between two immiscible phases. In the mixture, logP is a single parameter that combines several effects such as all non-covalent interactions, solvation and entropy components into a single component. The lipophilicity parameter is accessible for all five (5) studied molecules in the calculation software such as ChemSketch, ChemDraw and Molinspiration.
3. Results and Discussion
3.1. Calculs DFT
Understanding the delicate interplay between a molecule’s geometric structure, its properties, and the sensitivity to computational tools is crucial for reliable prediction and interpretation of its chemical behavior [23] . Density Functional Theory (DFT) has demonstrated several advantages over other computational methods when calculating compounds of organic or inorganic origin. It also provides low computational cost and reliable results to quantum chemical molecular.
To gain deeper insights into the molecular structure and reactivity potential of benzothiazoles and their derivatives, density functional theory (DFT) calculations were employed at the B3LYP-6-31+G (d, p) level, providing detailed information about electron distribution and chemical behavior. Density functional theory (DFT) with the B3LYP functional was employed within Gaussian 09 software to optimize molecular geometry, analyze frontier molecular orbitals (FMOs), calculate global reactivity descriptors, and map the molecular electrostatic potential (MEP). Additional visualization and analysis were facilitated by GaussView 6.0. The 6-31+ G (d, p) basis set was used for all calculations. Molecular boundary orbitals (HOMO and LUMO) represent key factors in understanding and predicting the molecule’s behavior, as they directly influence its electronic properties. Within a conjugated system, the HOMO energy and spatial distribution serve as markers for nucleophilic reactivity, whereas the LUMO energy and distribution indicate potential electrophilic reactivity, guiding the prediction of potential reaction sites [24] .
3.2. Reactivity Study
3.2.1. Global Reactivity Indices
Further delving into the molecule’s chemical nature, parameters like chemical potential (μ), global electrophilicity (ω), global hardness (η), and softness (s) offer complementary perspectives on its stability and reactivity, enriching our understanding [25] . Molecules with larger energy gaps are generally less susceptible to induced charge separation compared to those with smaller gaps, influencing their interactions with external fields.
The analysis of HOMO and LUMO energies, as employed in this study, is a reliable tool for evaluating the electron-donating and electron-accepting properties of molecules, as supported by [26] [27] . Additionally, this approach allows us to identify molecules with lower kinetic stability due to smaller HOMO-LUMO gaps. Quantum chemical calculations, specifically by determining the HOMO- LUMO energy gap, offer a reliable approach to assess the chemical reactivity of the molecule. Smaller gaps, indicating easier electron transfer, are associated with higher polarizability and enhanced reactivity, while larger gaps signify greater stability and lower reactivity [27] . By studying the properties of the HOMO and LUMO, we can gain valuable information about a molecule’s susceptibility to reactions, its capacity for excited-state behavior, and its propensity to interact with other molecules, all of which contribute to understanding its potential biological activity [28] . The calculated energy values for the FMOs (EHOMO, ELUMO) and their energy gaps, as well as the global reactivity descriptors in the absence of a solvent, are mentioned in Table 1. Figure 1 presents the structure of the molecules studied.
![]()
Table 1. Energy gaps (ΔE), Electronegativity (χ), chemical potential (μpot), hardness (η), softness (S) and electrophile number (ω) calculated in gase phase at B3LYP/6-31+G (d, p) level.
Substitution at position 2 of the ring leads to a progressive increase in the energy gap (ΔE), suggesting significant effects of substituents on the electronic structure. Substitution in position 2 therefore reduces the reactivity of benzothiazole (BTH). The lowest value of ΔE (0.1841 eV) is obtained with 2-SCH3_BTH, which remains the most reactive and least stable molecule, while 2-OH_BTH has the highest value of ΔE (0.2058 eV) and is the least reactive and most stable (Table 1). Beyond the parameters already discussed, several established descriptors like the chemical potential (μ), electrophilicity index (ω), hardness (η), and softness (s) provide complementary insights into the molecule’s intrinsic reactivity within the broader molecular system [24] . The polarizability of a molecule is inversely related to its HOMO-LUMO energy gap. Molecules with large gaps, known as ‘hard’ molecules, require significant energy to promote electrons from occupied to unoccupied orbitals, making them less susceptible to polarization compared to “soft” molecules with smaller gaps. BTH has a higher electronegativity value (χ = 0.1511 eV) than other compounds, so it is the best electron acceptor. 2-SCH3_BTH has the lowest chemical hardness value (η = 0.153 eV) and the highest softness (s = 6.5372 eV−1) of all the molecules. Thus 2-SCH3_BTH appears to be the most reactive of all the compounds under investigation. Additionally, the value of the electrophilicity index (ω = 0.1375 eV) of BTH indicates that it is the most electrophilic.
3.2.2. Dipole Moment
The dipolar force is a good indicator of the reactivity of a chemical compound. By measuring the dipole moment, we gain insights into the molecule’s overall shape and how its electron density is distributed. It’s worth noting that even-carbon molecules typically possess larger dipole moments compared to odd-carbon ones, demonstrating an “odd-even effect” in this property.
Analysis of the table shows that the dipole moments of the molecules studied have positive values (Table 2), so they are all polar. We have thus established the sequential classification below in ascending order of polarity: 2-OH_BTH < 2-SCH3_BTH < 2-SH_BTH < BTH < 2-NH2_BTH It follows from this order
![]()
Table 2. Values of calculated dipole moment in gas phase at B3LYP/6-31+G (d, p) level.
aStructure-activity relationship by the QSAR method of antibiotic molecules. bTheoretical Study of the Mechanism of Corrosion Inhibition of Carbon Steel in Acidic Solution by 2-aminobenzothaizole and 2-Mercatobenzothiazole. cEffective protection for copper corrosion by two thiazole derivatives in neutral chloride media: Experimental and Computational Study.
that the compound 2-OH_BTH is the least polar and 2-NH2_BTH is the most polar compound. The compound 2-NH2_BTH with the greatest dipole moment is considered the most water-soluble and the lowest organic solubility. The 2-OH_BTH molecule is the least water-soluble and and the highly miscible in a variety of organic liquids.
3.2.3. Lipophilic
A compound’s ability to interact with liquids, known as lipophilicity, plays a crucial role in determining its pharmacological effects (pharmacodynamics), its circulation in the body (pharmacokinetics) and its potential for toxicity. This technique offers a reliable way to evaluate the molecule’s organic phase affinity, directly linked to the strength and nature of intermolecular interactions with organic solvent molecules, such as hydrogen bonding and dipole-dipole interactions [29] [30] . This study seeks to establish a comprehensive understanding of the lipophilicity profile of benzothiazole and its derivatives through both experimental and theoretical approaches.
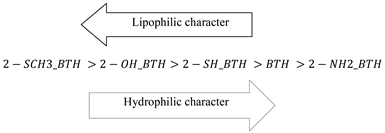
The lipophilic properties (LogP) of the target compounds were calculated using ChemSketch, ChemDraw and Molinspiration. Positive values of lipophilicity have been obtained for the series of studied molecules. This shows that the studied molecules are all lipophilic (hydrophobic). As Table 3 shows, BTH, 2-SH_BTH, 2-OH_BTH and 2-SCH3_BTH have possessed the most favorable partition coefficient values, indicating a strong tendency to reside in lipid-rich phases as compared to aqueous environments. Furthermore, the least value was found for 2-NH2_BTH. These results are in great agreement with experimental results. Taking into account the results of two software packages, namely, ChemSketch and ChemDraw we established the following sequential ranking in order of decreasing lipophilicity.
![]()
Table 3. Calculated values of lipophilicity with different software packages.
aHANSCH, C et al. (1995). bBrownlee BG et al.; Environ Toxicol Chem 11: 1153-1168 (1992). cMeylan WM, Howard PH; J Pharm Sci 84: 83-92 (1995). dACD Laboratories, www.acdlabs.com.
3.3. Stability Study
Using B3LYP/6-31+G (d, p) optimized geometries, we determined the thermodynamic parameters of the molecules (Table 4). Interestingly, all calculated standard enthalpies of formation were negative. Thermodynamic analysis based on the measured enthalpy and free enthalpy values, both found to be negative, confirms the exothermic and spontaneous nature of the reaction under the chosen experimental conditions. In the realm of thermodynamics, a negative entropy value signifies a system transitioning towards greater order and organization, with decreased randomness and complexity. Thus, the formation of all the molecules occurs spontaneously, releasing heat and decreasing the system’s overall disorder. The obtained parameters at the B3LYP/6-31+G (d, p) level provide strong evidence for the existence of the explored benzothiazole series under realistic conditions (298.15 K and 1 atm).
3.4. Molecular Electrostatic Potential (MEP)
Analyzing the MEP surface, which maps the electrostatic potential around a molecule, allows us to predict the relative reactivities of different positions towards nucleophilic and electrophilic reagents based on their charge preferences. We employed density functional theory (DFT) calculations at the B3LYP/6-31+G (d, p) level of theory on the optimized molecular structures to generate the molecular electrostatic potential (MEP) surfaces of the compounds. By employing a color-coded scheme, we can gain insights into the molecular recognition process, analyze hydrogen bonding patterns, and readily identify potential electrophilic and nucleophilic reaction sites. Electrostatic potential of the molecule surfaces of benzothiazole and its compounds were calculated and their MEP diagrams are shown in Figure 2.
The MEP model is used to deduce reactive sites on compounds [31] that can potentially be considerd as electrophilic or nucleophilic attack sites. This particular visualization method, frequently employed in the field, effectively reveals potential sites for intermolecular interactions and preferred reaction locations within a molecule [32] [33] . These MEPs provide several comprehensive data on
![]()
Table 4. Thermodynamic data for formation of benzothiazole and derivatives optimised derivatives at B3LYP/6-31+G (d, p) level.
the distribution of electrostatic charges on the optimised ground state geometries of our molecules, as illustrated in Figure 2. Careful analysis of Figure 2 shows that the compounds charge density distribution is more uniform in our molecules, leading to a less polarised ground state. The spatial distribution of electrostatic potential across the studied molecules is visualized in Figure 2. The colour code of the substances is ranging from −6.529e−2 to 6.529e−2 including the others. The blue and red colours in the PEM substances indicate respectively a nucleophilic site which is a rich region in terms of electron, and an electrophilic site which is a poor region in terms of electron. The polarisation effect is clearly visible in the compounds. In benzothiazole, the intense blue zone, or electropositive site, was found to be located mainly around carbon atom (C7). The N13 atom in benzothiazole also has a large red zone (negative electron site). This observation is also valid for the other compounds where the N13 atom has a large red zone and is therefore the nucleophile. As for the electrophilic sites, they vary from one compound to another. For 2-SH_BTH, the intense blue zone around the S14 atom, 2-NH2_BTH (N14), 2-OH_BTH (O14) and 2-SCH3_BTH (C3 and C15).
3.5. NBO (Natural Bonding Orbital) Analysis
NBO analysis offers a powerful tool for unraveling intermolecular interactions and bond characteristics, while also serving as a convenient platform for investigating charge transfer and conjugation within molecular systems. Natural Bond Orbital (NBO) analysis offers valuable insights into hyperconjugative interactions within a molecule by revealing electron delocalization patterns and providing quantitative measures of interaction energies. The valence occupancies anti-bonds unresolvable signal deviations from an idealised localised Lewis compound, signifying true “delocalisation effects”. Stability in the system arises when electron density flows from the donor orbital to the acceptor orbital through these interactions. The Natural Bond Orbital (NBO) method allows for a quantitative analysis of this donor-receptor interaction through the second-order perturbation interaction energy (E (2)), providing insights into the strength and nature of the electron transfer. This calculated energy value serves as a prediction for the magnitude of the off-diagonal elements within the NBO Fock matrix, quantifying the interaction strength between specific occupied orbitals. From the second-order perturbation approach [34] [35] , it can be inferred. For studied compounds, analysis Natural Bond Orbital (NBO) was calculated with DFT/B3LYP/631+G (d, p), level in gaseous and aqueous media with the continuous polarisable conducting-type solvation model (CPCM). The following tables summarize the electron-donating and accepting orbitals involved in key interactions, along with their associated stabilization energies, as determined through second-order perturbation theory analysis of the NBO Fock matrix.
3.5.1. Benzothiazole (BTH)
Valeurs in Table 5 demonstrates that a stabilizing interaction occurs between the electrons in a C-C bond and the anti-C-C bond within the ring due to strong intramolecular hyperconjugation. This interaction stabilizes the nuclear framework of the molecule. Our molecule exhibits diverse electronic interactions, involving both stabilizing (bonding) and destabilizing (anti-bonding) interactions between occupied and unoccupied orbitals, including non-bonding lone pairs. These interactions lead to Intramolecular Charge Transfer (ICT), which stabilises the various molecules of the systems. In the case of BTH, the highest stabilisation energy is about 26.11 kcal/mol. This value is observed at the level of interaction between the free electron pairs LP (2) (S14) → π* (C7-N13). The π → π* transition was observed at the interaction between C5 → C6 with a stabilisation energy of 20.38 kcal/mol. Concerning the solvent (water) the large stabilisation energy value is observed for the interaction between LP (2) (S14) → π* (C7-N13) with a value of 26.97 kcal/mol. Also, the π → π* transition was observed at the interaction between C5 → C6 and C1 → C2 with a stabilisation energy of 20.59 kcal/mol. Our analysis revealed that lone pair interactions from sulfur atoms S14 and the anti-bonding character of π (C7-N13) bonds contribute to the molecule’s stability. Interestingly, water as the solvent influences the second-order interaction energy (E (2)) differently for various transitions, as detailed in (Table 6).
![]()
Table 5. Fock matrix for benzothiazole in gas phase analyzed using second-order perturbation theory based on Natural Bond Orbital (NBO) analysis.
![]()
Table 6. Fock matrix for benzothiazole in aqueous phase (water) analyzed using second- order perturbation theory based on Natural Bond Orbital (NBO) analysis.
3.5.2. Benzothiazole Derivatives in the Gas Phase
We employed second-order perturbation theory to analyze and quantify the energetic interactions between non-Lewis Natural Bond Orbitals (NBOs) of the acceptor and Lewis-type NBOs of the donor molecule. Natural bond orbital (NBO) analysis was employed to assess electron distribution across individual atoms within the studied molecules. This analysis enabled the estimation of stabilization energy (E (2)) associated with electron delocalization between specific donor (i) and acceptor (j) NBOs, as detailed in reference [36] . The following tables present the calculated perturbation energies for various donor-acceptor interactions among benzothiazole derivatives.
The results of the NBO analysis compiled in Table 7 show that the bonding orbitals LP (2) S13 for 2-SH_BTH, LP (1) N14 (2-NH2_BTH), LP (2) S14 (2-SCH3_BTH) and LP (2) O14 (2-OH_BTH) have the largest E (2) values. They participate as donors and the π* (C7 - N12) bonding orbitals as acceptors respectively in the gas phase with stabilisation energies that range from 24 to 55 kcal/mol (Table 7), that result in intramolecular charge transfer, which leads to stabilisation of the molecules. Unlike π → π* (often), or LP (1) → σ* transitions, and σ → σ* interactions, they are due to a weak donor-acceptor interaction, resulting in low stabilisation energy values. It can therefore be concluded that the interactions between LP (1) N14 and LP (2) S13 and S14 strongly contributed to the stabilisation of the studied molecules as evidenced by the high values of their stabilisation energy.
3.5.3. Benzothiazole Derivatives in the Aqueous Phase
In aqueous phase (water), It is observed that LP2 (S13) → π* (C7-N12) (2-SH_BTH, 2-NH2_BTH) with stabilization energies 26.61, 25.47 kJ/mol, LP2 (S14) → π* (C7-N12) (2-SCH3_BTH) with maximum stabilization energy 27. 52 kJ/mol and LP2 (O14) → π* (C7-N12) (2-OH_BTH) has the maximum stabilisation 39.21 kJ/mol compared to all molecules stabilisation energy (Table 8). Our analysis revealed a close resemblance between the intramolecular interactions observed in the two studied benzothiazole derivatives and those predicted for isolated molecules in the gas phase, suggesting minimal environmental influence on these interactions within the studied systems. In contrast to the stronger interactions observed previously, the maximum second-order interaction energy (E2) associated with intermolecular charge transfer in this context remains relatively low, not exceeding 8 kcal/mol. In σ (C1-N12) → σ* (C7-S14) (2-SH_BTH, 2-SCH3_BTH), σ (C1-N12) → σ* (C7-N14) (2-NH2_BTH) and σ (C1-N12) → σ* (C7-O14) (2-OH_BTH). The σ → σ* interaction has a smaller energy E (2) than LP → π*or σ*. It ranges from 5.95 kcal/mol to 7.91 kcal/mol in vacuum, from 5. 92 kcal/mol to 7.89 kcal/mol in water (Table 8). Regarding the large contribution of the stabilisation energy E (2) for LP (2) → π* interactions compared to that of π donor orbitals with π* acceptor, we can conclude that polar covalent bond interactions contribute more to molecule stabilisation than van der Waals interactions.
![]()
Table 7. Second-order perturbation theory analysis conducted on the natural bond orbital (NBO)-based Fock matrix for benzothiazole derivatives in gas phase.
![]()
Table 8. Fock matrix for benzothiazole derivatives in aqueous phase (water) analyzed using second-order perturbation theory based on Natural Bond Orbital (NBO) analysis.
4. TD-DFT Analysis of the Absorption Spectra of Benzothiazole and Its Derivatives
Table 9 presents calculated absorption spectra (TD-B3LYP/6-31+G (d, p), gas phase) and the most likely electronic transitions for benzothiazole and its derivatives in terms of transition probability. According to (Table 9), the primary electronic transitions in all studied molecules involve excitations from the HOMO (highest occupied molecular orbital) to the LUMO (lowest unoccupied molecular orbital). These HOMO-LUMO transitions are responsible for the observed λ wavelengths. As shown in (Table 9), the maximum absorption peak obtained from benzothiazole in vacuum is located at 264.65 nm, which is in disagreement with the literature [37] . This disagreement can be explained by the choice of method. The λ wavelengths of most of the benzothiazole derivatives are greater than 250 nm, indicating a violet shift in absorption relative to the BTH nucleus. In comparison with the absorption peak corresponding to the S1 excited state of BTH, that of 2-SH_BTH, 2-SCH3_BTH and 2-NH2_BTH exhibits a bathochromic shift of 3.67 nm, 8.5 nm and 8.41 nm that of 2-OH_BTH exhibits a hypsochromic ultraviolet shift of 7.92 nm respectively, indicating that the substitution position of the different groups may have an effect on the excited state properties of BTH. The characteristic electronic spectrum allows for
![]()
Table 9. Excitation energies (ΔEexcit) in electronvolts (eV), wavelengths (λ) in nanometers (nm), oscillator strength (f), and electronic transitions of the absorption maxima of benzothiazole and its derivatives calculated at B3LYP/6-31+G (d, p) level in gas phase.
![]()
Table 10. Excitation energies (ΔEexcit) in electronvolts (eV), wavelengths (λ) in nanometers (nm), oscillator strength (f), and electronic transitions of the absorption maxima of benzothiazole and its derivatives calculated in aqueous phase at B3LYP/6-31+G (d, p) level.
clear differentiation: transitions with wavelengths longer than 270 nm are assigned to n → π electronic transitions, while those shorter than 270 nm are attributed to n → σ excitations.
We employed TD-DFT calculations at the B3LYP/6-31G (d) level, starting from the optimized ground state structures, to elucidate the electronic transitions responsible for the observed spectra of the complexes in aqueous solution (CPCM model [38] ). All predictions for the UV-vis spectra are summarised in (Table 10). For BTH, the wavelengths are virtually identical. The same observation is made for excitation energies. We note that water has no impact on the electronic transitions. Beyond this, we note that water modifies (increases or decreases) certain transitions without affecting the excitation energies.
5. Conclusions
This work involved studying the reactivity and stability of benzothiazole and its derivatives using DFT method. Amongst the studied compounds, 2-hydroxybenzothiazole emerged as the most reactive according to our reactivity analysis, which employed various global reactivity indices derived from quantum chemical calculations. The moment of the dipole calculated numbers with in the gas phase show that our compounds are all polar and a calculation of the lipophilicity confirms that our compounds are lipophilic (hydrophobic). Based on thermodynamic data obtained at 298.15 K, the formation and persistence of the benzothiazole series and its derivatives were predicted, suggesting their potential existence under ambient conditions.
The studied compounds exhibited distinct MEP profiles, readily revealing potential attack sites for electrophiles (negative potential zones) and nucleophiles (positive potential zones) through visual inspection of the MEP maps. NBO analysis provided detailed insights into the interplay between electron delocalization, hyperconjugation interactions, and intramolecular charge transfer, ultimately contributing to the stabilization energy of the studied molecules. This refined Lewis structure, based on natural orbital analysis, contributes to improved understanding of the molecule’s chemical behavior and properties by accurately representing its electronic state. Utilizing NBO analysis, we identified strong intramolecular hyperconjugation interactions within the molecules, which likely contribute to their remarkable stability in both gas and aqueous phases. To achieve a comprehensive understanding of environmental effects, we adopted a comparative TD-DFT approach, analyzing the electronic absorption spectra of our molecules in both gas and aqueous phases. Using computational methods, we identified potential absorption bands between 264.65nm and 277.23 nm for all molecules, accompanied by significant charge transfer. This finding, achieved using our chosen methodology, suggests the involvement of n → π* et n → σ* transitions in the observed electronic spectrum.