We report results from
ab-initio, self-consistent density functional theory (DFT) calculations of electronic, transport, and related properties of chromium disilicide (CrSi
2) in the
hexagonal C40 crystal structure. Our computations utilized the Ceperley and Alder local density approximation (LDA) potential and the linear combination of atomic orbitals (LCAO) formalism. As required by the second DFT theorem, our calculations minimized the occupied energies, far beyond the minimization obtained with self-consistency iterations with a single basis set. Our calculated, indirect band gap is 0.313 eV, at room temperature (using experimental lattice constants of a = 4.4276
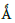
and c = 6.368
). We discuss the energy bands, total and partial densities of states, and electron and hole effective masses. This work was funded in part by the US Department of Energy, National Nuclear Security Administration (NNSA) (Award No. DE-NA0003679), the National Science Foundation (NSF) (Award No. HRD-1503226), LaSPACE, and LONI-SUBR.